We’ve all heard about the race to find a COVID-19 vaccine, but have you ever wondered how scientists were able to design treatments so quickly? The answer lies in how researchers were able to decode the virus’s molecular structure despite having no prior knowledge of its protein makeup. With no existing templates to work from, Ab Initio Modeling became the key tool in predicting the 3D structure of these unfamiliar proteins. Researchers were able to uncover how the virus worked at a molecular level, and ultimately, create life-saving vaccines by modeling of COVID-19 virus proteins.
Ab Initio Modeling has already proven to be indispensable across many scientific fields, offering solutions where traditional techniques fall short. While methods like homology modeling and X-ray crystallography rely on known structural templates, Ab Initio Modeling does not require any prior knowledge, making it the go-to method for predicting structures of proteins that have no known counterparts. The very nature of Ab Initio Modeling has made it crucial for industries such as drug discovery, biotechnology, and environmental science.
This blog aims to shed light on how this powerful method works, why it’s gaining prominence, and its vital role in advancing modern science. Whether you’re a student, researcher, or industry professional, understanding Ab Initio Modeling is key to staying at the forefront of innovations in health, medicine, and beyond.
| Interesting Facts: Protein structure‑prediction techniques (including ab initio methods) are being leveraged to uncover new therapeutic targets when no template structures exist. Ab initio modelling supports the design of novel enzymes and proteins for industrial use (e.g., bio‑catalysis, environmental remediation) by predicting structures from scratch. |
What is Ab Initio Modeling?
Ab Initio Modeling is a computational method used to predict the three-dimensional structure of a protein directly from its amino acid sequence, without relying on any existing structural templates. Essentially, it’s like creating a 3D map of a protein based solely on the chemical properties of its components, atoms, bonds, and interactions, rather than copying from known structures. This approach allows scientists to explore and understand proteins that have never been studied before, offering new insights into their function and role in biological processes.
Apart from Ab Initio Modeling, there are two other common methods for protein structure prediction: homology modeling and X-ray crystallography. However, homology modeling is limited to proteins with known structural templates and relies on sequence similarity, while X-ray crystallography requires experimental data, which can be time-consuming and difficult to obtain for certain proteins. Due to these limitations, Ab Initio Modeling has become the go-to method for predicting the structures of novel proteins that lack known templates, as it works entirely from the amino acid sequence without any pre-existing structural data.
| Quick Fact: Ab Initio Modeling has seen dramatic improvements in accuracy and efficiency, thanks to the integration of deep learning techniques for protein structure prediction. By using deep neural networks, tools like DeepFold predict spatial restraints more accurately, enhancing protein folding simulations. This approach has shown up to 44.9% higher accuracy compared to traditional methods and increased folding speed by 262 times. |
Ab Initio Modeling: The Process
Ab Initio Modeling addresses a fundamental question in biology: how does a protein fold into its unique three-dimensional shape? By simulating the protein’s natural folding process, it provides critical insights into the physical forces, such as atomic interactions and energy landscapes, that drive the formation of its final structure. This ability to model the intrinsic folding process is what truly sets Ab Initio Modeling apart.
Step 1: Sequence Input
The first step in Ab Initio modeling begins with the sequence input, where the amino acid sequence of the target protein is provided as the core input. This sequence serves as the foundational data for the entire modeling process. It is obtained from sources like genomic sequencing, protein identification through mass spectrometry, or databases like UniProt.
Once retrieved, the sequence is used to set up the necessary energy functions and simulations that will drive the subsequent folding process. Unlike template-based methods, Ab Initio modeling doesn’t rely on any known structures but instead builds the 3D structure entirely from the sequence itself.
This step ensures that the protein is represented as a simplified structure, typically focusing on the backbone first, which will be further refined as the simulation progresses. The sequence input step effectively sets the stage for accurate folding simulations by preparing the protein’s basic structure for energy calculations and conformational sampling in later steps.
Step 2: Energy Calculation
The Energy Calculation step in Ab Initio modeling plays a crucial role in determining the stability and feasibility of the generated protein structures. This step involves calculating the interactions between atoms using a defined energy function, which models the physical forces acting on the protein during folding. Traditionally, physics-based energy functions, such as those derived from classical force fields like AMBER, CHARMM, and GROMOS, are used to simulate these atomic interactions. These force fields account for various energy terms, including bond lengths, angles, torsion angles, van der Waals forces, and electrostatic interactions. The energy calculation is guided by these potentials to identify the most stable conformation of the protein from a large set of possible structures (decoys).
Modern practices in energy calculation have evolved significantly, with advances in molecular dynamics (MD) simulations coupled with these force fields providing insights not only into the folded structure but also into the folding process itself. While fully quantum mechanical simulations are still computationally impractical for large proteins, hybrid methods combining physics-based potentials and knowledge-based approaches are increasingly used. For example, methods like ROSETTA and TASSER utilize knowledge-based energy functions to further refine low-resolution models and improve the folding predictions by incorporating data from the protein’s sequence, secondary structure, and fragments of known protein structures. This combination of physics-based energy functions and knowledge-based methods significantly improves the accuracy and efficiency of protein structure prediction, making the process more feasible and reliable for larger proteins.
Step 3: Conformational Sampling
In Ab Initio modeling, conformational sampling is the process where the model explores a wide range of possible folding configurations. During this step, various algorithms like Monte Carlo simulations or molecular dynamics (MD) simulations are employed to sample different structures based on the energy function. These methods explore the conformational space by repeatedly generating new structures, refining them, and checking their energy states to find the most thermodynamically favorable conformations.
The goal of conformational sampling is to identify the lowest-energy states that are closest to the native structure of the protein. Efficient sampling is crucial, as an exhaustive search of all possible configurations would be computationally infeasible. Techniques like simulated annealing or genetic algorithms are used to optimize the search process, enabling the identification of near-native structures from a large pool of decoys.
Step 4: Energy Minimization
Once a set of potential conformations has been sampled, the next step is energy minimization, where the model is refined to achieve the lowest possible energy state. This process involves adjusting the structure to minimize any unfavorable interactions, such as steric clashes or poor bond angles, that may have resulted during the sampling phase. The energy function used in this step evaluates the stability of the structure by calculating the interaction energies between atoms, such as van der Waals forces, electrostatic interactions, and hydrogen bonding.
Energy minimization algorithms iteratively adjust the atomic coordinates to reduce the overall energy of the structure. These adjustments are done using optimization techniques like the steepest descent or conjugate gradient methods. The goal is to reach a stable conformation that corresponds to the thermodynamically most favorable structure, ensuring that the model is as close to the native state as possible. This step is critical in eliminating unrealistic features that may have been introduced during the sampling phase, leading to a more accurate final model.
Step 5: Structure Optimization
Following energy minimization, structure optimization aims to refine the model further by improving its overall stability. This step ensures that the protein adopts its most stable 3D conformation by adjusting the side-chain positions and fine-tuning bond angles, torsions, and other atomic interactions. Structure optimization may involve the use of molecular dynamics simulations or Monte Carlo methods to explore the conformational space more thoroughly. The goal is to obtain a model that closely mimics the natural folded structure of the protein, minimizing any steric clashes or unfavorable configurations that may have been overlooked during earlier stages.
During this phase, the model undergoes iterative refinement to achieve a configuration where energy is minimized across the entire structure, especially focusing on the protein’s flexibility and local interactions. By exploring alternative configurations and optimizing atomic positions, the process ensures that the final model represents the most likely and thermodynamically stable form of the protein under physiological conditions.
Step 6: Validation
Once the structure has been optimized, validation ensures the accuracy and reliability of the predicted model. This step involves comparing the generated structure to known experimental data, if available, or evaluating it using computational techniques such as root-mean-square deviation (RMSD), Ramachandran plot analysis, and other structural metrics. Validation tools can assess if the predicted model aligns with expected biophysical properties and if it contains any errors, such as unfavorable bond angles or clashes.
The final validated model is then submitted to protein databases like the Protein Data Bank (PDB) or other relevant repositories, where it can be accessed by the scientific community for further research. The validation process is critical in ensuring that the model not only satisfies theoretical predictions but also holds up to experimental scrutiny, making it a valuable resource for researchers in drug discovery, structural biology, and other applications.
Ab Initio Modeling Methods
Ab initio modeling techniques are essential in predicting the three-dimensional structures of proteins without relying on any prior template structures. These methods focus on simulating the folding process of proteins directly from their amino acid sequences, using various computational approaches. Below is an overview of some of the primary methods used in ab initio modeling:
1. Physics-Based Simulations
Physics-based simulations are widely used in ab initio modeling as they form the foundation for the energy calculations and conformational optimization of protein structures. These simulations are employed throughout the entire process of structure prediction, from the initial model generation to refinement, because they provide detailed insight into molecular interactions, such as van der Waals forces, electrostatic interactions, and bond lengths. However, while physics-based simulations are integral to many stages, they are not typically used for large-scale, high-resolution folding due to their computational intensity when dealing with large systems.
This method is most suitable for cases where accurate energy calculations are crucial, such as minimizing structural energy in low-resolution models. It is especially effective when dealing with small to medium-sized proteins and when a detailed energy landscape is needed for modeling interactions. Physics-based simulations are also commonly used for structure refinement after initial folding has occurred through other methods, providing further energy optimization.
2. Monte Carlo Simulations
Monte Carlo (MC) simulations are among the most commonly used methods in ab initio modeling. They are popular due to their simplicity, efficiency, and ability to explore large conformational spaces quickly. This makes them highly effective in situations where a broad search of possible folding states is required but detailed dynamics are not as important. Their relatively lower computational cost makes them the method of choice for many initial foldings or decoy generation tasks.
Monte Carlo simulations are most suitable for tasks that require rapid exploration of conformational space, particularly during the early stages of protein folding. They are ideal for large proteins where the goal is to sample a vast number of potential conformations rather than simulating detailed molecular motion. MC methods are also highly useful in optimization processes, such as finding low-energy conformations quickly in systems where time-dependent behavior is not crucial. MC simulations are commonly employed in high-throughput settings, where multiple models need to be generated and evaluated efficiently.
3. Molecular Dynamics
Molecular Dynamics (MD) simulations are less commonly used than MC simulations in ab initio modeling due to their high computational cost and the significant time requirements involved in simulating protein folding over long timescales. While MD offers detailed, time-resolved simulations of protein motion, it is typically reserved for cases where a deeper understanding of protein dynamics is needed. The high cost in terms of computational resources means that MD simulations are generally only used when necessary.
Molecular dynamics is most suitable for situations where the time-dependent behavior of proteins needs to be simulated. This includes cases where understanding the folding pathway, intermediate states, and dynamic motions of the protein is crucial. MD is ideal when studying small to medium-sized proteins (less than 100–200 residues), and it excels in cases where protein function is tied to dynamic motion, such as in enzymatic activity or protein-ligand interactions. MD simulations are also used in refinement stages to assess the final stability and flexibility of the predicted structure.
Case Study: Ab Initio Modeling in Action
Problem:
When the SARS-CoV-2 virus emerged, scientists were faced with the challenge of understanding its molecular structure, particularly the spike protein responsible for entering human cells. With no existing templates in protein databases, predicting its structure was crucial for designing vaccines and therapeutics. Traditional structure determination methods, such as X-ray crystallography or cryo-EM, were not feasible for this novel virus, highlighting the importance of computational methods like Ab Initio Modeling.
What Was Done:
Ab Initio Modeling was employed to predict the spike protein’s 3D structure directly from its amino acid sequence. The sequence of input given into computational models allowed for simulations and energy-based calculations to generate various potential structures. Monte Carlo simulations and Physics-Based Simulations were particularly valuable in exploring conformational states and optimizing the predicted structure.
Steps Involved:
- Sequence Input: The spike protein’s amino acid sequence was used as the starting point for structure prediction.
- Energy Calculation: Computational methods calculated the energy of different conformations based on force fields.
- Conformational Sampling: Monte Carlo simulations and other physics-based approaches explored multiple potential 3D structures.
- Structure Optimization: The most stable structure was identified and refined using molecular dynamics simulations.
- Validation: The predicted structure was compared with known structures of related viruses to ensure its biological relevance.
Importance of Ab Initio Modeling:
The inability to use existing templates made Ab Initio Modeling the only viable method for predicting the spike protein’s structure. By predicting this structure from scratch, researchers could identify key regions for therapeutic intervention. The modeling helped in understanding how the spike protein interacts with the ACE2 receptor, enabling the development of targeted vaccines and antiviral drugs.
Outcome:
The 3D structure of the SARS-CoV-2 spike protein significantly accelerated vaccine development. Researchers were able to design vaccines like Pfizer-BioNTech and Moderna by targeting specific regions of the spike protein, preventing the virus from entering human cells. The predicted structure also played a role in the development of therapeutic antibodies aimed at blocking the viral entry mechanism.
Key Takeaways:
This case study highlights the power of Ab Initio Modeling in solving urgent global health challenges. The ability to predict protein structures without templates was key in rapidly advancing SARS-CoV-2 vaccine development, demonstrating the vital role of computational modeling in modern drug discovery and disease management.
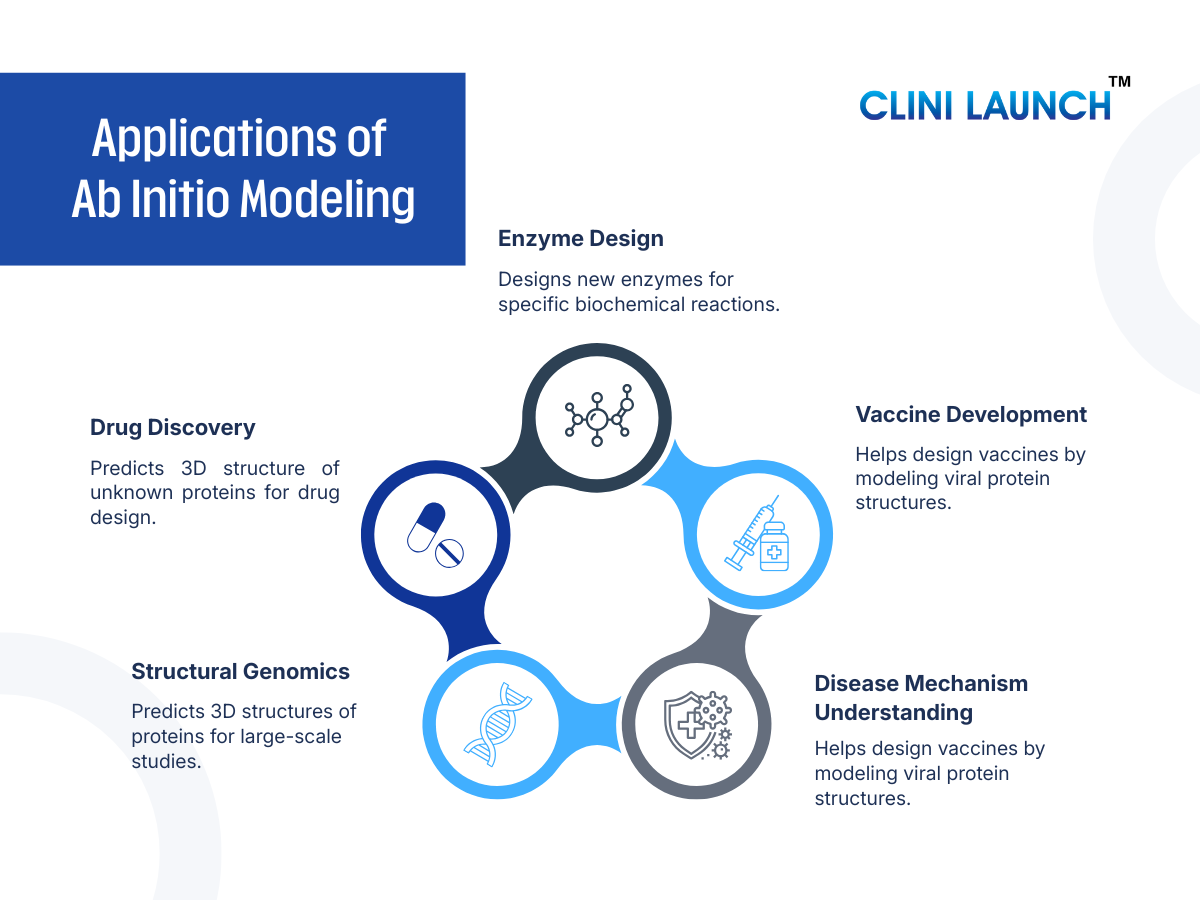
Applications of Ab Initio Modeling
1. Drug Discovery
Drug discovery primarily involves identifying molecules that can interact with specific biological targets to treat diseases. When structures of targets (such as proteins) are not known, Ab Initio modeling can be critical in predicting their 3D structure from just the amino acid sequence.
During the COVID-19 pandemic, researchers used Ab Initio modeling to predict the structure of the SARS-CoV-2 spike protein. This prediction helped in understanding how the virus binds to human cells, guiding the design of vaccines like the Pfizer-BioNTech and Moderna vaccines. This approach helped accelerate vaccine production and antiviral drug screening.
Ab Initio methods were employed to model the spike protein’s 3D structure, even before experimental methods like X-ray crystallography or cryo-EM could be applied. This was essential in the early stages of vaccine development, as the spike protein was identified as the key target for vaccine design. By predicting its structure, researchers were able to develop vaccines faster, improving the global response to the pandemic.
2. Enzyme Design
Ab Initio modeling is especially useful when no natural enzyme template exists, and researchers need to design a protein with a desired function from scratch. Enzyme design involves creating new enzymes or optimizing existing ones for specific biochemical reactions.
Enzyme design for biofuel production has seen significant advancements through Ab Initio modeling. For instance, researchers have designed enzymes capable of breaking down complex biomass into sugars, which can be used in biofuel production. Traditional enzyme design was limited by available templates, but Ab Initio methods allowed for the creation of entirely new enzymes based on the sequence of amino acids alone.
Using Ab Initio methods, researchers can predict the structure of novel enzymes with high specificity and functionality. This includes modeling the enzyme’s active site and its interaction with substrates, helping in the design of more efficient biocatalysts.
Rothlisberger et al. Used Ab Initio methods to design a novel enzyme, successfully demonstrating its ability to catalyze a reaction that was previously only achievable through traditional chemical methods. This enzyme was created de novo, illustrating the potential of Ab Initio in enzyme design.
3. Vaccine Development
Vaccine development relies on understanding the structure of viral proteins to create an immune response. Ab Initio modeling is particularly important when the structure of the virus is unknown or difficult to determine.
The development of the Ebola virus vaccine was greatly accelerated by the use of Ab Initio modeling to predict the structure of the Ebola glycoprotein, a key target for vaccine development. Researchers modeled the 3D structure of the glycoprotein from its amino acid sequence, allowing them to identify the binding sites for neutralizing antibodies. This structural prediction led to the development of a vaccine that is now used in outbreak areas.
4. Disease Mechanism Understanding
Understanding disease mechanisms at the molecular level is critical for developing effective treatments. Ab Initio modeling is particularly useful in studying diseases linked to novel or poorly characterized proteins, where no known templates exist.
Alzheimer’s Disease research has benefited from Ab Initio modeling in understanding the role of amyloid-beta peptides, which are implicated in the formation of plaques in the brain. By modeling these peptides’ 3D structures, researchers can identify the mechanism of plaque formation and design drugs to inhibit it.
Liu et al. (2015) used Ab Initio modeling to study the aggregation process of amyloid-beta peptides in Alzheimer’s Disease. Their work revealed key structural features that were targeted for drug development.
Ab Initio modeling was used to predict the structure of amyloid-beta peptides and their aggregation patterns. This insight helped in the design of drugs that can inhibit plaque formation or disrupt the aggregation process.
5. Structural Genomics
Structural genomics aims to determine the 3D structures of proteins on a large scale, particularly for proteins with unknown structures. Ab Initio modeling plays a significant role when no homologous protein templates are available.
Tina et al. (2007) developed an Ab Initio method for protein structure prediction that contributed significantly to structural genomics efforts. Their method was applied to several previously uncharacterized proteins, expanding our understanding of protein functions in the human genome.
In structural genomics projects like the Human Genome Project, Ab Initio modeling has been used to predict the structures of thousands of proteins with unknown sequences, aiding in the cataloging of protein functions.
Ab Initio methods were employed to predict the 3D structures of proteins encoded by genes with no known homologous structures, allowing researchers to assign functions to these uncharacterized proteins.
Conclusion
Ab Initio Modeling has proven to be an indispensable tool in predicting protein structures, especially when no existing templates are available. By using just the amino acid sequence, it allows researchers to accurately model the 3D structures of proteins, enabling advancements in drug discovery, enzyme design, and vaccine development. The prediction of the SARS-CoV-2 spike protein is a prime example of how Ab Initio Modeling can accelerate scientific breakthroughs and help address urgent global health challenges.
Looking ahead, innovations such as AI-enhanced Ab Initio Modeling are set to further revolutionize the field. AI algorithms can optimize and refine computational methods, significantly improving accuracy and reducing simulation times. These advancements will not only enhance our understanding of complex proteins but also accelerate the development of targeted therapeutics, opening new doors for breakthroughs in biotechnology and medicine.
If exploring how scientists decode life from molecules to data sparks your curiosity, then the CliniLaunch’s Advanced Diploma in Bioinformatics is the perfect next step — where you’ll learn the very tools and techniques that bring orphan proteins to life.



