

Site monitoring in clinical trials is a critical control mechanism that ensures a study is conducted exactly as approved. It exists to confirm that clinical trial activities follow the protocol, meet regulatory compliance requirements, and uphold ethical standards throughout the trial lifecycle.
At the center of this process is the Clinical Research Associate (CRA), who serves as the operational link between the sponsor and the investigator site. Through structured site monitoring visits, the CRA verifies that patient safety is protected, trial data is accurate and traceable, and essential documents are properly maintained.
Errors in trial conduct or documentation, if left undetected, can compromise data integrity, delay regulatory submissions, or raise serious compliance concerns during inspections. For this reason, site monitoring in clinical trials is not a routine formality. It is a safeguard that supports clinical trial oversight and ensures that studies generate reliable and credible results.
This blog explains what site monitoring in clinical trials involves, the different types of clinical trial monitoring used in practice, and the step-by-step site monitoring process followed from site setup to study close-out, supported by a real-world case example.
What is Clinical Trial Monitoring?
Clinical trial monitoring is a systematic process used to ensure that a clinical study is conducted, recorded, and reported in accordance with the approved protocol, Good Clinical Practice (GCP), regulatory requirements, and ethical principles.
In practical terms, clinical trial monitoring functions as an ongoing quality control activity. It focuses on verifying that participant rights and safety are protected, that adverse event reporting is accurate and timely, and that trial data reflects what actually occurred at the site. This includes reviewing source documents, confirming protocol compliance, and ensuring that deviations are identified, documented, and addressed appropriately.
An important aspect of site monitoring in clinical trials is its role in maintaining regulatory inspection readiness. Well-monitored sites are more likely to demonstrate compliance because essential documents, such as the Investigator Site File and Trial Master File, are kept current and complete. Through regular monitoring, sponsors gain confidence that the trial is being conducted as intended and that the data generated can withstand regulatory review.
Types of Clinical Trial Monitoring
Clinical trials differ in complexity, risk profile, geographic spread, and data volume. For this reason, site monitoring in clinical trials is not performed using a single fixed approach. Instead, different types of clinical trial monitoring are applied based on study needs, regulatory expectations, and risk assessment outcomes.
Each monitoring method serves a specific purpose, and in practice, most trials use a combination rather than relying on only one approach.
On-Site Monitoring
On-site monitoring is the traditional and most direct form of site monitoring in clinical trials. In this approach, the Clinical Research Associate conducts monitoring visits by physically visiting the investigator site. These visits allow the CRA to directly observe trial conduct and verify that study procedures are being followed exactly as described in the protocol.
During an on-site monitoring visit, the CRA reviews source documents to perform source data verification, checks informed consent documentation, assesses adverse event reporting, and evaluates drug accountability and storage conditions. Essential documents maintained in the Investigator Site File are also reviewed to confirm regulatory compliance.
Because the CRA is present at the site, on-site monitoring allows for immediate clarification of issues and direct interaction with site staff. However, it is time-intensive and contributes significantly to monitoring-related costs in clinical trials.
Remote Site Monitoring
Remote site monitoring allows the CRA to conduct monitoring activities without physically visiting the site. Instead, monitoring is performed using secure electronic systems such as Electronic Data Capture platforms, electronic Trial Master Files, and Clinical Trial Management Systems.
Through remote monitoring, the CRA can review trial data, track protocol deviations, assess documentation completeness, and follow up on monitoring findings in a timely manner. This approach improves efficiency, reduces travel requirements, and allows more frequent data review compared to traditional on-site visits.
Remote site monitoring is particularly effective for ongoing data checks and document reviews. However, it has limitations when it comes to verifying physical processes, investigational product handling, and site facilities.
Centralized Monitoring
Centralized monitoring is a data-focused approach in which study data from all participating sites is reviewed centrally by the sponsor or contract research organization. Using statistical tools and data analytics, centralized monitoring helps identify trends, outliers, missing data, or unusual patterns that may indicate quality or compliance issues.
This method supports early risk detection across multiple sites and enhances overall clinical trial oversight. Centralized monitoring is especially useful in large, multi-center studies where consistent site-level issues may not be immediately visible through individual monitoring visits.
While centralized monitoring strengthens trial-level oversight, it does not replace site-level verification and is typically used alongside on-site or remote monitoring.
Risk-Based Monitoring (RBM)
Risk-based monitoring is an approach that focuses monitoring efforts on the aspects of a trial that pose the greatest risk to participant safety and data integrity. Instead of applying the same level of monitoring to all sites and activities, RBM uses predefined risk assessments and ongoing data evaluation to guide monitoring intensity.
Under risk-based monitoring, high-risk processes such as informed consent, primary endpoint data, and safety reporting receive greater attention, while lower-risk activities may be monitored less frequently. This approach allows resources to be used more effectively while maintaining regulatory compliance.
RBM typically combines centralized monitoring, remote monitoring, and targeted on-site monitoring as part of a structured site monitoring plan.
Hybrid Monitoring
Hybrid monitoring combines elements of on-site and remote monitoring. In this approach, critical activities such as source data verification, informed consent verification, and drug accountability are performed during on-site visits, while routine data reviews and document checks are handled remotely.
Hybrid monitoring provides a balanced approach, maintaining oversight of high-risk areas while improving efficiency. As clinical trials increasingly adopt digital systems, hybrid monitoring has become a widely used model in modern studies.
Comparison on types of clinical trial monitoring
| Monitoring Type | Where It Happens | Key Activities | Strengths | Limitations |
| On-Site Monitoring | At the clinical trial site | SDV, IP checks, IC review, facility observation | Most comprehensive; direct oversight | Time-consuming; travel cost |
| Remote Monitoring | Off-site (online review) | EDC review, document checks, communication | Fast, cost-effective, continuous access | Limited ability to verify physical processes |
| Centralized Monitoring | Sponsor/CRO central systems | Data analytics, trend checks, anomaly detection | Early detection of deviations across sites | Does not replace site-level verification |
| Risk-Based Monitoring (RBM) | Combination of methods | Risk assessment, targeted checks | Optimizes resources; focuses on critical risks | Requires strong data systems & planning |
| Hybrid Monitoring | Mix of on-site + remote | Critical tasks on-site, routine tasks remote | Balanced efficiency and quality | Coordination needed between monitoring types |
Clinical Trial Monitoring Step-by-Step Process
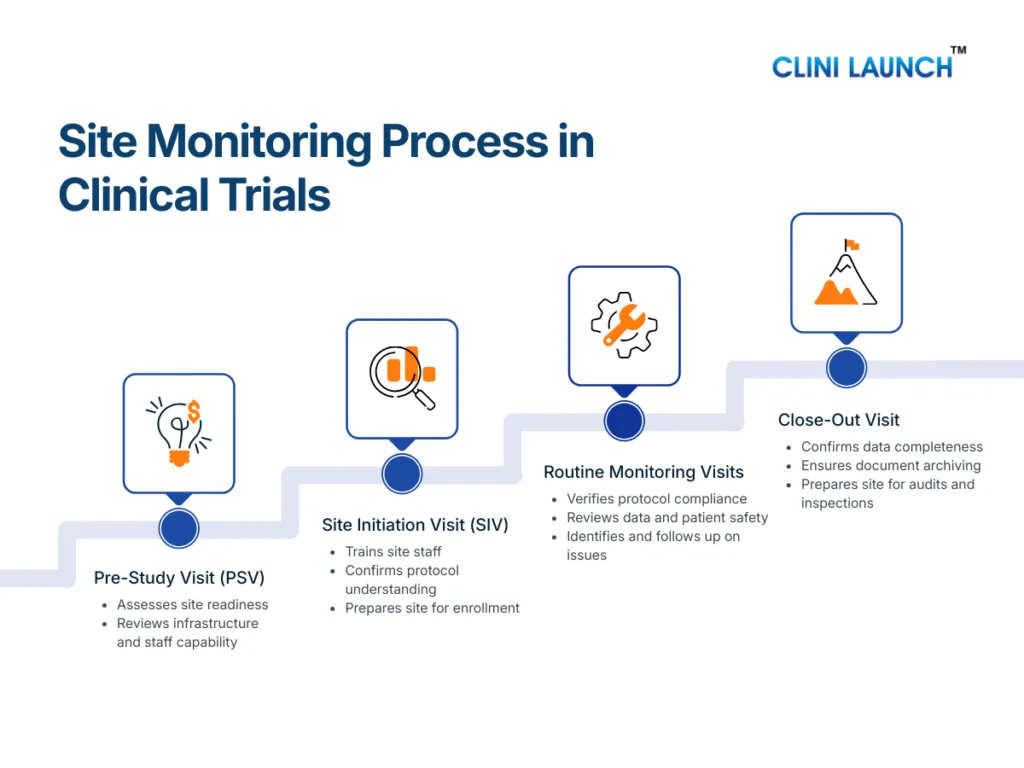
The site monitoring process in clinical trials follows a structured sequence of visits conducted across the study lifecycle. Each stage serves a distinct purpose, but together they ensure protocol compliance, patient safety monitoring, and data integrity from study start to closure.
The Clinical Research Associate is responsible for planning, executing, documenting, and following up on these monitoring activities as part of ongoing clinical trial oversight.
Pre-Study Visit (PSV): Assessing Site Readiness
The Pre-Study Visit is conducted before a site is authorized to participate in a clinical trial. Its primary objective is to assess whether the site is capable of conducting the study in accordance with the protocol and regulatory requirements.
During the PSV, the CRA evaluates the site’s infrastructure, including clinical facilities, investigational product storage areas, and data handling systems. The qualifications and experience of the investigator and site staff are reviewed to ensure they are appropriate for the study. The CRA also assesses whether the site can manage essential documents, adverse event reporting, and patient records in a compliant manner.
This visit plays a preventive role. By identifying gaps early, the CRA can guide the site on corrective actions before trial initiation, reducing the risk of compliance issues later in the study.

Site Initiation Visit (SIV): Preparing the Site for Trial Conduct
The Site Initiation Visit formally marks the transition from site preparation to active trial conduct. At this stage, the CRA ensures that the site fully understands the study requirements and is ready to enroll participants.
During the SIV, the CRA reviews the approved protocol in detail with the site team, explaining study objectives, eligibility criteria, visit schedules, and safety reporting expectations. Training is provided on informed consent procedures, data entry into the EDC system, and handling of investigational products. The CRA also confirms that all essential documents are in place and that the site monitoring plan is clearly understood.
A well-executed SIV establishes consistency in trial conduct and reduces the likelihood of protocol deviations during enrollment and follow-up.
Routine Monitoring Visits: Maintaining Ongoing Oversight
Routine monitoring visits are conducted at regular intervals throughout the trial and represent the core of site monitoring in clinical trials. These visits allow the CRA to verify that the study continues to be conducted as approved.
During routine monitoring visits, the CRA reviews participant eligibility, confirms that informed consent was obtained correctly, and monitors patient safety through adverse event reporting. Data entered into the CRF or EDC system is compared with source documents as part of source data verification. The CRA also reviews the Investigator Site File to ensure that essential documents remain current and complete.
Any issues identified during these visits are documented as monitoring findings. The CRA works with the site to resolve these issues and, where necessary, supports corrective and preventive actions to prevent recurrence.
Close-Out Visit: Completing Site Responsibilities
The Close-Out Visit is conducted once all trial activities at the site have been completed. The purpose of this visit is to ensure that the site has fulfilled all protocol, regulatory, and documentation requirements before the study is formally closed.
During the close-out visit, the CRA confirms that all data queries have been resolved and that the study data is complete and accurate. Essential documents are reviewed to ensure proper archiving, and regulatory compliance is verified. The CRA also ensures that investigational products are returned or destroyed according to the protocol and applicable regulations.
This final monitoring stage ensures that the site is prepared for audits or inspections and that the study can progress confidently toward analysis and reporting.
Case Study on Clinical Trial Monitoring
How a Routine Monitoring Visit Prevented Data Integrity Issues
This case illustrates how effective site monitoring in clinical trials directly protects data quality and scientific validity.
During a routine monitoring visit, the Clinical Research Associate observed that a study participant scheduled for a Day 5 visit arrived nearly two hours later than planned. Despite the delay, the site staff proceeded with dosing and pharmacokinetic (PK) or pharmacodynamic (PD) sample collection without documenting the deviation or questioning its impact.
At first glance, the situation appeared operationally minor. However, in clinical trials involving PK or PD assessments, sample timing is critical. Even small deviations can significantly affect data interpretation.
Identification of the Issue
When the CRA later reviewed the source documents, a serious discrepancy became evident. The visit was documented as if it had occurred exactly according to the protocol-defined schedule. There was no record of the delayed arrival, no protocol deviation reported, and the PK or PD sample times were recorded based on planned rather than actual collection times.
This meant that the recorded data did not accurately reflect what occurred at the site. Because PK analyses depend on precise timing relative to dosing, the inaccurate documentation had the potential to distort the participant’s concentration profile and compromise the scientific integrity of the dataset.
CRA Action and Follow-Up
Having directly observed the deviation, the CRA escalated the issue to the Principal Investigator and the sponsor. The CRA ensured that the source documents were corrected to reflect the actual visit and sample collection times and that a formal protocol deviation was documented.
In addition, the CRA supported corrective and preventive actions. These included targeted protocol and GCP retraining for the site staff and the introduction of a checklist to reinforce real-time documentation during critical visits. These actions were aimed at preventing similar issues in future visits.
Outcome and Key Learning
Because the issue was identified and addressed promptly, inaccurate PK data was prevented from entering the final analysis. The CRA’s intervention preserved the reliability of the study data and supported regulatory compliance.
This case highlights the value of routine monitoring visits and demonstrates how vigilant site monitoring helps protect patient safety, data integrity, and overall trial credibility. It also reinforces why site monitoring is a critical safeguard rather than a procedural formality.
Conclusion
Site monitoring is one of the most operationally critical functions in clinical trials. It is where protocol design, regulatory expectations, and real-world site execution intersect. Understanding how site monitoring works—across different monitoring types and visit stages—provides a practical view of how clinical trials are actually controlled and safeguarded.
For individuals looking to enter clinical research, this knowledge is not optional. Roles such as Clinical Research Coordinator, Clinical Trial Assistant, and Clinical Research Associate all require a working understanding of site monitoring, protocol compliance, essential documentation, and patient safety oversight.
At CliniLaunch Research Institute, the PG Diploma in Clinical Research is designed to build this exact operational understanding. The program focuses on real clinical trial workflows, including site monitoring processes, CRA responsibilities, regulatory compliance, and inspection readiness—preparing learners to function confidently in entry-level and growing clinical research roles.
For those aiming to move from academic knowledge to industry-ready capability, structured training aligned with real trial operations makes the difference.
Frequently Asked Questions (FAQs)
Who decides how site monitoring will be conducted in a clinical trial?
The sponsor, often with input from the CRO, defines the site monitoring plan. This plan outlines the monitoring approach, visit frequency, and responsibilities based on study risk and complexity.
Is site monitoring mandatory for all clinical trials?
Yes. Some form of site monitoring is required for all interventional clinical trials. The method may vary, but oversight of site activities is always expected by regulators.
Can a clinical trial run without on-site monitoring?
In some studies, monitoring may rely more on remote or centralized methods. However, critical activities such as informed consent and investigational product handling usually still require on-site verification at some stage.
What happens if monitoring issues are not corrected?
Unresolved monitoring findings can lead to protocol deviations, regulatory observations, delayed approvals, or rejection of trial data during inspections.
How are monitoring findings documented?
Monitoring findings are recorded in a monitoring visit report. The site is required to respond, and corrective and preventive actions are tracked until closure.
Does site monitoring replace audits or inspections?
No. Site monitoring is a routine oversight activity. Audits and regulatory inspections are independent reviews conducted by sponsors or authorities to assess overall compliance.
Is site monitoring only the responsibility of the CRA?
The CRA leads monitoring activities, but investigators and site staff are responsible for correcting issues and maintaining compliance at the site.
Why do beginners struggle with site monitoring concepts?
Most academic programs focus on theory, while site monitoring involves operational decision-making, documentation control, and real-time risk management that are learned through practice.







