India’s pharmaceutical and clinical research ecosystem plays a central role in both national healthcare delivery and global drug development. As of FY 2023–24, India’s pharmaceutical market was valued at around USD 50 billion, with approximately 20 % of the world’s generic medicines supplied from Indian companies to countries across the globe. Indian firms also supply a major share of global vaccines, reinforcing the sector’s strategic importance.
This growth has set the foundation for careers of lakhs of professionals across the country. From drug development and clinical trials to safety monitoring and medical technology analytics, companies across these domains are actively recruiting freshers into meaningful roles. Whether you’re a life sciences graduate, a data enthusiast, or curious about regulatory and safety careers, many of these opportunities are driven by the rapid expansion of clinical research companies in India.
The clinical research industry in India is becoming deeply integrated with global development pipelines, and major clinical trial companies increasingly rely on Indian talent and infrastructure.
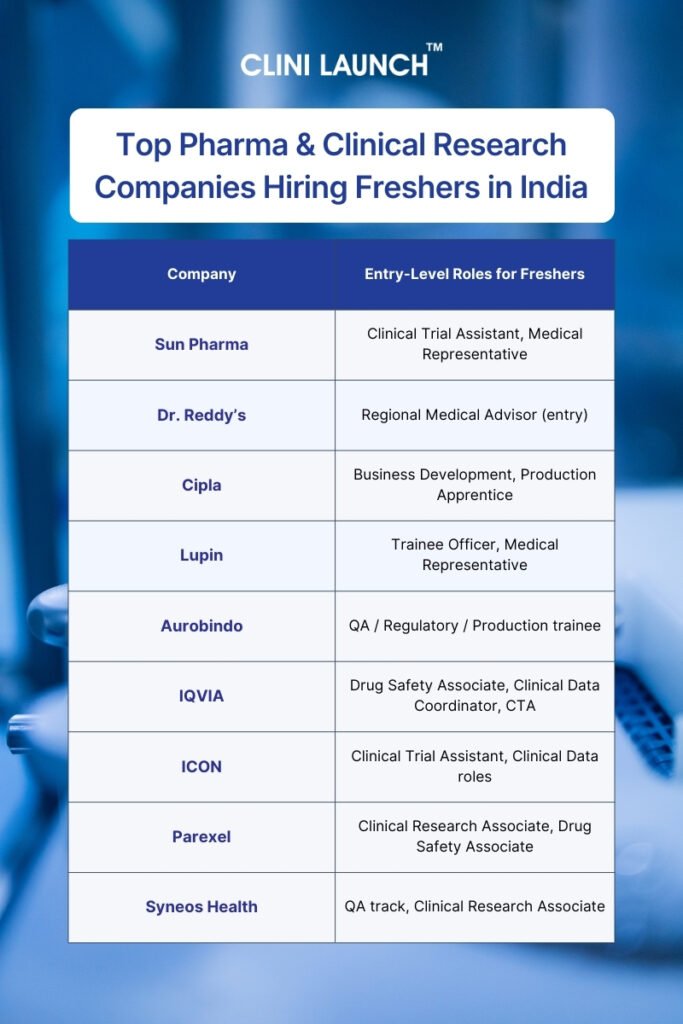
In this blog, we’ll highlight the Top 14 Pharmaceutical, Clinical Research and related companies hiring freshers in India. For the benefit of aspirants, the companies are categorized across core domains like pharmaceuticals, clinical research, pharmacovigilance, and data management, so you can discover where the jobs are and how to tap into them.
| Overview of the Medical Industry and Career Opportunities |
The medical industry includes pharmaceutical companies, clinical research organizations, drug safety teams, data management groups, and medical device companies that develop, test, monitor, and regulate medicines and healthcare technologies.
In India, this industry is rapidly growing and offers freshers non-patient-facing careers in clinical research, pharmacovigilance, data analytics, quality, and regulatory roles across global companies.
|
Scope for Careers in Pharmaceutical and Clinical Research Companies in India
In India, pharmaceutical exports reached USD 30.47 billion in 2024–25, reflecting robust manufacturing and global demand for Indian medicines and vaccines. At the same time, the global contract research organisation (CRO) services market was valued at about USD 85.54 billion in 2024 and is projected to grow significantly through the next decade, driven by the increasing outsourcing of clinical trials and R&D activities. These growth trends indicate sustained expansion across pharmaceutical manufacturing, clinical operations, pharmacovigilance, and data management functions. As companies scale their global delivery capabilities, they continue to create structured entry-level opportunities for fresh graduates in regulated, process-driven roles across India.
Why is the scope for freshers is expanding in Indian Pharma and CRO Space?
- Increased global outsourcing of clinical trials and R&D to India
Global pharmaceutical sponsors increasingly outsource clinical trials, data management, and regulatory operations to India due to cost efficiency, skilled talent availability, and regulatory experience. This has strengthened India’s position as a preferred destination for CRO-led research activities.
- Growth in generic drug exports and vaccine manufacturing
India’s strong manufacturing infrastructure and regulatory-approved facilities have boosted generic drug and vaccine exports to global markets. As production volumes rise, companies require trained professionals across quality, regulatory, safety, and operational roles.
- Rising demand for pharmacovigilance and regulatory compliance
Precise global safety monitoring requirements and evolving regulatory frameworks have increased demand for pharmacovigilance and compliance professionals. Continuous safety reporting and risk management have become core operational functions within pharma and CRO companies.
- Digital transformation in clinical data management and biostatistics
The adoption of electronic data capture (EDC), remote monitoring, AI-assisted analytics, and centralized trial management has expanded data-focused roles. Organizations now require professionals skilled in structured data handling, statistical programming, and data integrity oversight.
- Expansion of Global Capability Centers (GCCs) in India
Many multinational pharmaceutical and life sciences companies are expanding their Global Capability Centers in India to manage end-to-end research, safety, and data operations. These centers create consistent hiring pipelines for fresh graduates into standardized, globally aligned roles.
Advanced Diploma in
Clinical Research
Build practical, industry-aligned skills to work across real clinical trial environments. Learn how clinical studies are planned, conducted, documented, and monitored, with a strong emphasis on ethics, patient safety, and regulatory compliance throughout the trial lifecycle.
Top Pharmaceutical Companies Hiring Freshers in India
(Drug development & manufacturing)
These companies focus on discovering, developing, manufacturing, and distributing medicines and vaccines. Freshers are commonly hired into quality, regulatory, safety, production, and clinical support roles. Several pharma companies hiring freshers are building structured early-career pathways, and leading pharmaceutical companies in india continue expanding research and manufacturing investments. Competition among pharma companies hiring freshers in india has increased as global demand rises. Many of these employers collaborate closely with clinical research companies in india to support trial and safety programs.
1.Sun Pharmaceutical Industries
Sun Pharmaceutical Industries Ltd (Sun Pharma) is India’s largest pharmaceutical company and a globally recognized leader in generic and specialty medicines. It manufactures, develops, and markets a wide range of pharmaceutical formulations and Active Pharmaceutical Ingredients (APIs) across more than 100 countries, supported by 40+ manufacturing facilities worldwide. The company’s products cover major therapeutic areas such as cardiology, dermatology, neurology, diabetology, oncology, and more, making it a cornerstone of both Indian and global medicine supply chains.
Sun Pharma commands over 8% of the Indian pharmaceutical market, making it the top domestic pharma player by market share. Its global revenue is around US$ 6.2 billion, reflecting strong manufacturing, R&D, and export capabilities. The company also ranks among the largest specialty generic pharmaceutical firms in the world, trusted by healthcare professionals and patients for high-quality, affordable medicines.
Career Opportunities for Freshers in Sun Pharmaceutical Industries
Sun Pharma offers structured early-career opportunities and graduate trainee roles, especially in Clinical trial assistant, medical representative freshers. Fresh graduates can gain experience across cross-functional teams, exposure to regulated pharmaceutical processes, and mentorship from industry experts, making it a strong launchpad for careers in drug development and medical product operations.
Company Snapshot
| Category |
Details |
| Company Size |
~43,000+ employees globally (as of 2025) |
| What They Do |
Pharmaceutical formulations, generics, APIs, and specialty medicines |
| Headquarters |
Mumbai, India |
| Global Presence |
Medicines sold in 100+ countries worldwide |
| Notable Work |
Leading pharma market share in India with a wide therapeutic portfolio |
| Roles Hiring |
Clinical Trial Assistant, Medical Representative (Freshers) |
| Growth Focus |
Innovation in specialty generics and global expansion |
2. Dr. Reddy’s Laboratories
Dr. Reddy’s Laboratories Ltd is a major Indian multinational pharmaceutical company headquartered in Hyderabad. Founded in 1984, it manufactures and markets a broad portfolio of generics, branded generics, biosimilars, and APIs (Active Pharmaceutical Ingredients) across key global markets including India, the USA, Europe, and emerging regions. The company’s products span important therapeutic areas such as gastrointestinal, cardiovascular, oncology, respiratory, and dermatology, serving millions of patients worldwide.
In the fiscal year ending March 31, 2025, Dr. Reddy’s Laboratories reported an annual revenue of over ₹325 billion (~USD 3.9 billion), reflecting year-on-year growth driven by strong demand across markets and diversification of its product portfolio. The company also reported consistent revenue growth (~14% YoY) in the trailing twelve months, highlighting its strong performance and market resilience within the pharmaceutical sector.
Career Opportunities for Freshers in Dr. Reddy’s Laboratories
Dr. Reddy’s offers structured early-career roles and graduate trainee opportunities across Regional Medical Advisor. Fresh graduates can gain exposure to regulated drug development processes, global compliance standards, and cross-functional teams, making it a strong platform to build a career in pharmaceutical operations and drug lifecycle management.
Company Snapshot
| Category |
Details |
| Company Size |
~27,000+ employees globally |
| What They Do |
Generics, branded generics, biosimilars, APIs & OTC products |
| Headquarters |
Hyderabad, India |
| Global Presence |
USA, Europe, India & emerging markets |
| Notable Work |
Broad therapeutic portfolio with consistent revenue growth |
| Roles Hiring |
Regional Medical Advisor |
| Growth Focus |
Expansion of biosimilars and global generics pipeline |
3. Cipla Limited
Cipla Limited is one of India’s leading multinational pharmaceutical companies, founded in 1935 and headquartered in Mumbai. The company is known for its extensive portfolio of over 1,500 medicines across many therapeutic categories and its global presence spanning more than 80 countries. Cipla focuses on respiratory, anti-infective, urology, cardiology, and chronic disease portfolios, and it continues to expand through strategic launches and strong demand for its products in India and overseas.
Cipla reported revenue of approximately ₹27,267 crore (around USD 3.25 billion) in FY2025, with consistent double-digit growth in key markets and strong performance in respiratory and chronic care segments. The company also ranks among the top four pharmaceutical companies in India by revenue and holds a strong presence in high-growth markets across Africa and North America.
Career Opportunities for Freshers in Cipla Limited
Cipla recruits fresh graduates into roles such as Business Development, Apprenticeship in Production API, offering exposure to regulated pharmaceutical workflows and opportunities to work on large-scale manufacturing and global compliance projects. Its broad product portfolio and global operations make it a great launchpad for careers in drug development and regulated operations.
Company Snapshot
| Category |
Details |
| Company Size |
~30,000+ employees globally |
| What They Do |
Generics, APIs & therapeutic product portfolios |
| Headquarters |
Mumbai, India |
| Global Presence |
80+ countries |
| Notable Work |
Wide therapeutic portfolio with robust revenue growth |
| Roles Hiring |
Business Development, Apprenticeship in Production API |
| Growth Focus |
Respiratory and chronic disease portfolios; global market expansion |
4. Lupin Limited
Lupin Limited is a major Indian multinational pharmaceutical company headquartered in Mumbai, known for producing a wide range of generic medicines, complex generics, and active pharmaceutical ingredients (APIs). Founded in 1968, Lupin has grown into one of the top pharmaceutical players in India and the world, with its products sold in over 100 countries and a strong presence in key markets including India and North America.
In FY25, Lupin’s India business generated revenues of INR 75,773 million, accounting for about 34 % of its global turnover, and the company ranked as the 8ᵗʰ largest firm in the Indian Pharmaceutical Market (IPM) by value. This reflects Lupin’s strong footprint across chronic therapeutic areas such as cardiology, respiratory, diabetes, and anti-infective medicines.
Career Opportunities for Freshers in Lupin Limited
Lupin offers early-career opportunities and graduate trainee roles across Trainee Officer microbiology, Medical Representative. Fresh talent can gain hands-on experience in regulated drug development processes, quality systems, and global compliance frameworks, making it a great launchpad for careers in pharmaceutical operations, data analysis, and compliance.
Company Snapshot
| Category |
Details |
| Company Size |
~24,000+ employees globally (as of 2025) |
| What They Do |
Generic formulations, APIs, complex generics & therapeutics |
| Headquarters |
Mumbai, India |
| Global Presence |
Products sold in 100+ countries |
| Notable Work |
8th largest in IPM with a broad therapeutic portfolio |
| Roles Hiring |
Trainee Officer (Microbiology), Medical Representative |
| Growth Focus |
Expansion in chronic therapy segments and global markets |
5. Aurobindo Pharma
Aurobindo Pharma Ltd is a major Indian multinational pharmaceutical company headquartered in Hyderabad and recognized among the top global generic pharma manufacturers. It develops, manufactures, and markets a wide range of generic formulations, active pharmaceutical ingredients (APIs), biosimilars, and speciality products used in therapy areas such as cardiovascular, anti-infective, anti-diabetic, CNS, and more. The company exports its products to 150+ countries, making it one of India’s most internationally diversified pharmaceutical firms.
Aurobindo Pharma is ranked among the top 10 generic pharmaceutical companies in the world and has a strong global footprint across North America, Europe, Asia, Africa, and Latin America. In 2025, its consolidated revenues were substantial, reflecting its significant scale in formulations and API manufacturing, and it continues to expand its product portfolio and global partnerships.
Career Opportunities for Freshers in Aurobindo Pharma
Aurobindo offers structured graduate and trainee roles in areas such as quality assurance, regulatory compliance, production support, clinical operations, and supply chain analytics. Fresh graduates can benefit from hands-on experience in regulated pharmaceutical manufacturing, global compliance standards, and exposure to large-scale operations, making it an ideal launchpad for careers in the medical industry.
Company Snapshot
| Category |
Details |
| Company Size |
~40,750+ employees globally (2025) |
| What They Do |
Generic drugs, APIs, biosimilars, specialty formulations |
| Headquarters |
Hyderabad, India |
| Global Presence |
Products sold in 150+ countries |
| Notable Work |
Top 10 global generics company with a broad therapeutic portfolio |
| Roles Hiring |
Quality, Regulatory, Clinical, Production & Analytics Trainees |
| Growth Focus |
Expansion of global markets and diversified therapeutic portfolios |
Top Clinical Research & CROs Hiring Freshers in India
(Clinical trials & operations)
These companies manage and execute clinical trials for pharmaceutical and biotechnology firms. Freshers are commonly hired into clinical operations, data management, drug safety, and regulatory support roles. Many clinical research organizations in india work closely with global sponsors to manage multinational trials. A contract research organization in india may take responsibility for monitoring, safety reporting, and data delivery across regions. Several CRO companies in india now operate structured onboarding pipelines, and this model has made clinical research companies in india some of the largest recruiters of trained graduates. Below is a practical list of clinical research organizations frequently known for hiring beginners.
1.IQVIA
IQVIA Holdings, Inc. is one of the world’s largest clinical research organizations (CROs), integrating clinical trial services, advanced analytics, and real-world data to support drug development across the full lifecycle. The company works with pharmaceutical, biotechnology, and medical device organizations to manage Phase I–IV clinical trials, regulatory submissions, safety reporting, and real-world evidence generation across global markets.
IQVIA operates at the intersection of clinical research and data science, using large-scale healthcare datasets and technology platforms to improve trial efficiency, evidence generation, and regulatory decision-making worldwide.
In FY 2024, IQVIA reported revenue of USD 15.4 billion, reflecting its scale across clinical research, analytics, and life sciences services. The company also maintains one of the world’s largest healthcare data assets, with access to over 1.2 billion anonymized patient records, enabling advanced real-world evidence and outcomes of research used by regulators and life sciences companies globally.
Career Opportunities for Freshers in IQVIA
IQVIA offers structured entry-level and graduate roles across Drug Safety Associate, Clinical documentation specialist, Lab Center Project coordinator, Centralized Monitoring Assistant, Clinical Research coordinator, Clinical Data coordinator . Fresh graduates gain exposure to global clinical trials, regulated workflows, and data-driven research platforms, making IQVIA a strong starting point for careers in clinical research, trial operations, and life sciences analytics.
Company Snapshot
| Category |
Details |
| Company Size |
~88,000+ employees globally |
| What They Do |
Clinical trials (Phase I–IV), real-world evidence, analytics, safety & regulatory services |
| Headquarters |
Durham, North Carolina, USA |
| Global Presence |
Operations across 100+ countries |
| Notable Work |
Global clinical trial execution and real-world evidence platforms |
| Roles Hiring |
Drug Safety Associate, Clinical Documentation Specialist, Lab Center Project Coordinator,
Centralized Monitoring Assistant, Clinical Research Coordinator, Clinical Data Coordinator
|
| Growth Focus |
Advanced analytics, AI-enabled trials, decentralized and hybrid trial models |
2. ICON plc
ICON plc is a leading global clinical research organization (CRO) that provides outsourced clinical development, trial management, data analytics, and regulatory support services to pharmaceutical, biotechnology, and medical device companies. The company supports drug and therapeutic development across all phases of clinical research (Phase I–IV), helping clients speed up product development and bring safe, effective therapies to market. ICON also integrates advanced healthcare intelligence and digital solutions to optimize trial outcomes and evidence generation.
In 2024, ICON reported full-year revenues of approximately USD 8.28 billion, reflecting its global scale and leadership in clinical research services. ICON employs around 41,900 professionals across 106 locations in 55 countries, underscoring its expansive global footprint and ability to manage complex multinational trials.
Career Opportunities for Freshers in ICON PLC
ICON offers structured early-career opportunities and graduate roles in clinical trial assistant, clinical data management. Fresh graduates gain exposure to regulated workflows, real-world evidence studies, and global healthcare operations, ideal for building a career in research, analytics, and medical product support.
Company Snapshot
| Category |
Details |
| Company Size |
~41,900 employees globally (2024) |
| What They Do |
Clinical trials (Phase I–IV), data analytics, regulatory & development support |
| Headquarters |
Dublin, Ireland |
| Global Presence |
106 locations across 55+ countries |
| Notable Work |
Strong global CRO operations and healthcare intelligence solutions |
| Roles Hiring |
Clinical Trial Assistant, Clinical Data Management |
| Growth Focus |
Integrated research services, digital health, and data-driven trial insights |
3. Parexel
Parexel is a leading global clinical research organization (CRO) that supports pharmaceutical, biotechnology, and medical technology companies in planning, managing, and executing clinical trials (Phase I–IV) and related research functions. The company provides a wide range of services including clinical trial operations, regulatory strategy, data management, biostatistics, pharmacovigilance, and real-world evidence analysis, helping accelerate the development and global approval of new medicines and therapies.
Parexel operates with a global workforce of about 24,000 professionals, delivering clinical research support in numerous therapeutic areas including oncology, neuroscience, and infectious diseases — making it one of the world’s largest CROs. Additionally, the global clinical trials market was estimated at USD 48.2 billion in 2023 and is expected to grow to USD 73.2 billion by 2027, reflecting the growing scope and demand for CRO services like those Parexel provides.
Career Opportunities for Freshers in Parexel
Parexel provides opportunities for fresh graduates through roles such as Clinical Research Associate and Drug Safety Associate. These pathways introduce newcomers to clinical trial conduct, safety monitoring practices, and compliance-focused research environments while working with global teams. The experience supports steady career progression in clinical operations and pharmacovigilance.
Company Snapshot
| Category |
Details |
| Company Size |
~24,000+ employees globally (2025) |
| What They Do |
Clinical trial services (Phase I–IV), regulatory strategy, data & safety support |
| Headquarters |
Raleigh, North Carolina, USA |
| Global Presence |
Operates in 80+ countries (clinical sites & offices) |
| Notable Work |
Broad scope of clinical research services across therapeutic areas |
| Roles Hiring |
Clinical Research Associate, Drug Safety Associate |
| Growth Focus |
Global clinical trial delivery, regulatory support, and real-world evidence research |
4. Syneos Health
Syneos Health is a global clinical research organization (CRO) and biopharmaceutical solutions company that provides comprehensive support across clinical trials, data management, Regulatory strategy, and medical affairs. It partners with pharmaceutical, biotech, and medical device firms to help accelerate drug development, optimize trial outcomes, and bring safe, effective therapies to patients worldwide.
Syneos Health employs around ~28,000 professionals across more than 110 countries, serving clients in clinical research and commercial services. Its integrated model combines clinical trial management with data-driven insights and commercial expertise, reinforcing its position as one of the top CROs in the world.
Career Opportunities for Freshers in Syneos Health
Syneos Health provides entry pathways for fresh graduates in areas such as Quality Assurance and Clinical Research Associate tracks, Safety and PV coordinator. These opportunities help newcomers understand regulated research practices, study oversight expectations, and compliance-driven environments while working alongside global teams. The exposure builds a solid base for long-term careers in clinical research and trial operations.
Company Snapshot
| Category |
Details |
| Company Size |
~28,000+ employees globally (2025) |
| What They Do |
Clinical trials, data management, regulatory & commercial services |
| Headquarters |
Morrisville, North Carolina, USA |
| Global Presence |
Operations across 110+ countries |
| Roles Hiring |
Clinical Data Associate, Clinical Research Associate (Trainee Roles),
Quality Control Trainee, Drug Safety Associate
|
| Growth Focus |
Integrated research services and data-driven trial insights |
5. Labcorp Drug Development
Labcorp Drug Development (formerly Covance) is a leading global clinical research organization (CRO) and part of LabCorp’s biopharma services focused on clinical trials, drug development, safety testing, and regulatory support. It helps pharmaceutical and biotech companies accelerate therapies from early-stage research through late-phase clinical trials and approval, combining deep scientific expertise with extensive lab and trial infrastructure.
Labcorp Drug Development supports drug development services used in major clinical research programs worldwide. Its parent company, Labcorp, achieved annual revenue of USD 13.01 billion in 2024 and employs more than 70,000 people across 100+ countries, reflecting its global scale in diagnostics, drug development, and research services.
Career Opportunities for Freshers in Labcorp Drug Development
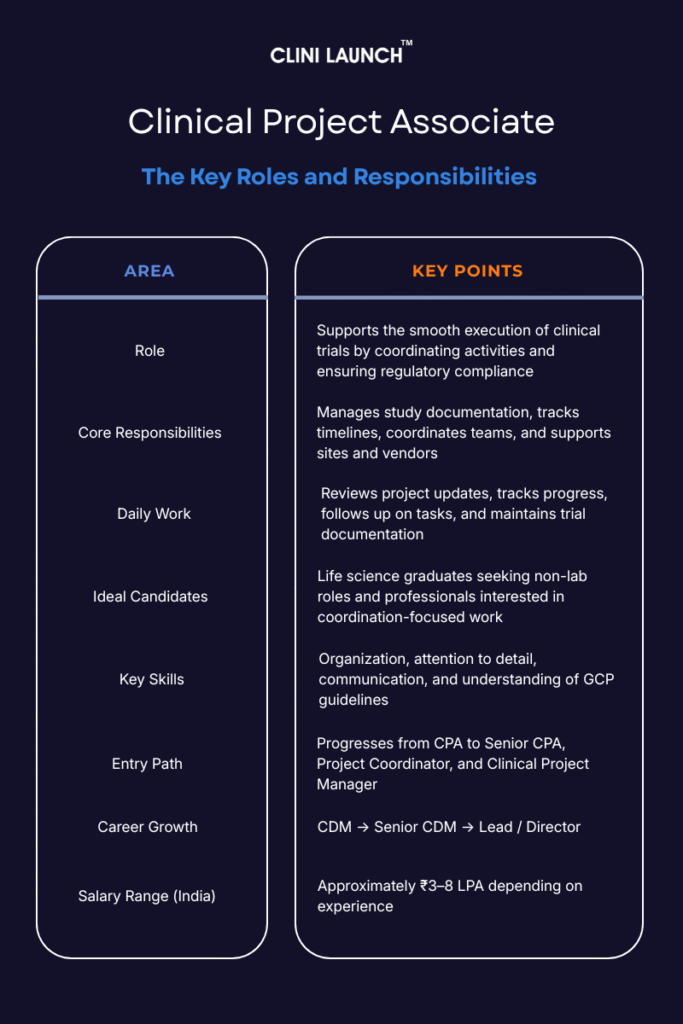
Labcorp Drug Development offers entry opportunities through roles such as Clinical Project Associate and Clinical Project Coordinator. These positions give fresh graduates exposure to global clinical workflows, trial documentation practices, and coordination activities within regulated research environments. The experience helps build a dependable base for long-term growth in clinical operations and pharmaceutical development.
Company Snapshot
| Category |
Details |
| Company Size |
Part of Labcorp (~70,000+ employees worldwide) |
| What They Do |
Drug development, clinical trials, safety & regulatory support |
| Headquarters |
Burlington, North Carolina, USA |
| Global Presence |
Services in 100+ countries |
| Notable Work |
Comprehensive drug development and clinical trial services |
| Roles Hiring |
Clinical Data Coordinators, QC Data Associates |
| Growth Focus |
End-to-end clinical trial delivery and regulatory excellence |
Top Pharmacovigilance & Drug Safety Companies Hiring Freshers in India
(Adverse event monitoring & compliance)
These companies focus on monitoring the safety of medicines and medical products during and after clinical trials. Freshers are commonly hired into drug safety operations, adverse events of case processing, safety data review, signal detection support, and regulatory compliance roles. Large drug safety companies india as well as global pharmacovigilance companies rely on standardized reporting systems. Many of them collaborate closely with clinical research companies in india to maintain continuous safety oversight.
1. Accenture Life Sciences
Accenture Life Sciences is the life sciences and healthcare services arm of Accenture, supporting pharmaceutical, biotechnology, and medical device companies across pharmacovigilance, drug safety, regulatory compliance, and clinical operations. The company helps organizations manage adverse event processing, aggregate safety reporting, signal detection support, regulatory submissions, and post-marketing surveillance on a global scale.
Accenture combines deep domain expertise in drug safety with advanced digital and analytics capabilities, enabling large pharmaceutical companies to modernize and scale their pharmacovigilance operations while meeting strict global regulatory requirements.
Accenture works with 9 out of the top 10 global pharmaceutical companies, delivering large-scale pharmacovigilance, regulatory, and compliance operations worldwide. With a global workforce of 700,000+ professionals, Accenture supports high-volume, regulated life sciences programs across multiple regions.
Career Opportunities for Freshers in Accenture Life Sciences
Accenture provides entry pathways through roles such as Clinical Data Associate and Drug Safety Associate. These positions introduce fresh graduates to clinical and safety data workflows, adverse event documentation, and compliance-focused processes within global delivery environments. Structured training and large-scale systems help build a reliable foundation for long-term growth in clinical research and pharmacovigilance.
Company Snapshot
| Category |
Details |
| Company Size |
700,000+ employees globally |
| What They Do |
Clinical Data Associate, Drug Safety Associate |
| Headquarters |
Dublin, Ireland |
| Global Presence |
Operations across 120+ countries |
| Notable Work |
Large-scale global PV and regulatory programs for top pharma clients |
| Roles Hiring |
Drug Safety Associates, PV Case Processors, Regulatory Support Trainees |
| Growth Focus |
PV automation, analytics-led safety operations, and compliance transformation |
2. Wipro Life Sciences
Wipro Life Sciences is the life sciences and healthcare services division of Wipro, supporting pharmaceutical, biotechnology, and medical device companies across pharmacovigilance, drug safety operations, regulatory compliance, and clinical support services. The company helps organizations manage adverse event processing, safety data management, aggregate reporting, regulatory documentation, and post-marketing surveillance at a global scale.
By combining drug safety domain expertise with technology, analytics, and process automation, Wipro enables life sciences organizations to run compliant, scalable pharmacovigilance operations aligned with global regulatory requirements such as FDA, EMA, and ICH guidelines.
Wipro reported gross revenue of approximately USD 10.8 billion in FY 2024, reflecting its scale as a multinational services company delivering regulated life sciences, pharmacovigilance, and compliance operations worldwide.
Career Opportunities for Freshers in Wipro Lifesciences
Wipro provides entry opportunities for fresh graduates through positions such as Drug Safety Associate and Drug Safety Analyst. These roles offer practical exposure to safety case handling, pharmacovigilance operations, regulatory documentation, and compliance-driven workflows. With structured training and global systems, newcomers gain the foundation needed to build long-term careers in drug safety.
Company Snapshot
| Category |
Details |
| Company Size |
230,000+ employees globally |
| What They Do |
Pharmacovigilance, drug safety operations, regulatory & compliance services |
| Headquarters |
Bengaluru, India |
| Global Presence |
Operations across multiple continents |
| Notable Work |
Large-scale global PV and regulatory programs |
| Roles Hiring |
Drug Safety Associates, Drug Safety Analyst |
| Growth Focus |
PV automation, analytics-driven safety operations, and compliance transformation |
Top Clinical Data Management & Biostatistics Companies ring Freshers in India
(Trial data, SAS, analytics)
These companies manage and analyze clinical trial data using SAS and statistical methods. Freshers typically enter through CDM, SAS programming, and biostatistics to support roles. Clinical data management companies play a critical role in preparing submission-ready information, and many of these opportunities later grow into specialized clinical data management roles. Sponsors and clinical research companies in india rely heavily on accurate datasets for regulatory approval.
1. Medpace
Medpace is a global full-service Clinical Research Organization (CRO) known for its fully in-house clinical data management and biostatistics teams. Unlike many CROs that outsource data functions, Medpace maintains integrated CDM, SAS programming, and biostatistics operations, allowing tighter control over trial data quality and regulatory readiness. The company has strong expertise in oncology, CNS, metabolic, and cardiovascular studies, where data accuracy and statistical rigor are critical.
Medpace reported annual revenue exceeding USD 2.0 billion, reflecting consistent growth driven by complex, data-intensive and late-phase clinical trials. Its fully in-house data and statistics model is widely regarded as a key differentiator, particularly for regulatory-focused studies and submission-ready datasets.
Career Opportunities for Freshers in Medpace
Medpace offers accessible entry-level pathways through positions such as Data Coordinator and Assistant Clinical Data Analyst. These roles help newcomers build hands-on familiarity with clinical trial datasets, data review practices, CDISC expectations, and regulator-ready workflows. The experience supports steady growth toward long-term careers in clinical data management and biostatistics.
Company Snapshot
| Category |
Details |
| Company Size |
~5,000+ employees globally |
| What They Do |
Clinical data management, biostatistics, SAS & trial analytics |
| Headquarters |
Cincinnati, Ohio, USA |
| Global Presence |
Operations across North America, Europe & Asia |
| Notable Work |
In-house CDM & biostatistics teams; late-phase and regulatory trials |
| Roles Hiring |
Data Coordinator, Assistant Clinical Data Analyst |
| Growth Focus |
Data-driven trials, regulatory submissions, and complex study analytics |
Advanced Diploma in
Biostatistics
Build strong foundations in statistical methods used in clinical research and healthcare studies. Learn how clinical trial data is analyzed, interpreted, and validated to support evidence-based decisions and regulatory submissions.
2. Quanticate
Quanticate is a global data-focused clinical research organization (CRO) specializing in clinical data management, biostatistics, SAS/statistical programming, and data analytics for pharmaceutical, biotech, and medical device companies. It delivers expert statistical services, clinical data capture, reporting, and real-world evidence analytics to support regulatory-ready clinical trial outcomes and submission-quality datasets.
Quanticate is recognized as a leading global data-centric biometric CRO, supporting comprehensive clinical data and biostatistics services that help sponsors transform raw clinical data into high-quality outputs for regulatory filings and scientific reporting across major therapeutic areas. The company operates multiple global offices — including locations in the UK, US, Canada, India, Poland, and South Africa — demonstrating its international footprint in life sciences data services.
Career Opportunities for Freshers in Quanticate
Quanticate provides structured opportunities in roles such as medical writing, data process associate, and statistical programming. These positions allow new professionals to work with regulatory-compliant clinical trial data, understand industry workflows, and collaborate with global research teams. The exposure builds practical knowledge, strengthens technical confidence, and creates a solid pathway toward long-term growth in clinical research, analytics, and biostatistics.
Company Snapshot
| Category |
Details |
| Company Size |
~280+ professionals globally (2025, est.) |
| What They Do |
Clinical data management, biostatistics, SAS/statistical programming & analytics |
| Headquarters |
Hitchin, United Kingdom |
| Global Presence |
UK, USA, Canada, India, Poland, South Africa |
| Notable Work |
Data capture, statistical analysis, and reporting for clinical trials |
| Roles Hiring |
Medical Writer, SAS/Statistical Programmers, Data Process Associate |
| Growth Focus |
Advanced analytics, real-world data, and evidence-based insights |
Conclusion
Breaking into the medical industry doesn’t require a clinical background or patient-facing experience. What it does require is an understanding of how medicines, trials, data, and safety systems work together behind the scenes. From pharmaceutical manufacturing and clinical research to drug safety, data management, and clinical technology platforms, this industry runs process, precision, and compliance, and that’s exactly where fresh graduates can build meaningful careers.
For many learners, this becomes the gateway to long-term pharma industry careers india.
For those starting out, the key is not just choosing a company but choosing the right domain and learning path. Each area highlighted in this blog offers a different kind of exposure, learning curve, and growth trajectory. Building a strong foundation in clinical research concepts, regulatory workflows, and industry practices can make entry into these roles far more structured and confident. Understanding how clinical research companies in india operate helps fresh graduates choose the right preparation strategy.
This awareness also improves how candidates target clinical research jobs india in a competitive market.
For freshers looking to prepare themselves for these opportunities, enrolling in a structured program like the CliniLaunch’s Advanced Diploma in Clinical Research can help bridge the gap between academic knowledge and real-world industry expectations making the first step into the medical industry clearer and more achievable. These skills are exactly what recruiters across clinical research companies in india look for in entry candidates.
FAQs
1. What is the medical industry?
The medical industry includes companies that develop, test, manufacture, monitor, and regulate medicines, vaccines, diagnostics, and medical devices before they reach patients.
2. How is the medical industry different from the healthcare industry?
The medical industry focuses on creating and validating medical products, while the healthcare industry focuses on delivering care to patients through hospitals and clinics.
3. Can freshers get jobs in the medical industry?
Yes. Many entry-level roles in clinical research, drug safety, data management, quality, and regulatory support are designed specifically for fresh graduates
4. What qualifications are required to enter the medical industry?
Most entry roles require degrees in life sciences, pharmacy, biotechnology, statistics, or related fields, along with basic understanding of regulated industry workflows.
5. Which medical industry domain is best for freshers?
Clinical research, pharmacovigilance, clinical data management, and medical coding are the most beginner-friendly domains due to structured processes and clear workflows.
6. What are the best entry-level roles in the medical industry?
Common entry-level roles include Clinical Research Coordinator, Clinical Data Associate, Drug Safety Associate, Medical Coder, and Regulatory Support Executive.
7. Is clinical research a good career for beginners?
Yes. Clinical research offers structured career paths, global demand, and non-patient-facing roles focused on documentation, compliance, and trial operations.
8. Can non-science graduates enter the medical industry?
Yes. Roles such as medical coding, data operations, and regulatory documentation are accessible to non-science graduates with industry-focused training.
9. What is the average salary for freshers in the medical industry in India?
Freshers typically earn between ₹2.5 and ₹5.5 LPA, depending on the role, domain, and company.
Yes. Continuous global demand for medicines, clinical trials, safety monitoring, and medical technologies makes the medical industry a stable long-term career choice.
10. Is the medical industry a stable career option?
Yes. Continuous global demand for medicines, clinical trials, safety monitoring, and medical technologies makes the medical industry a stable long-term career choice.