| Role of an IRB Coordinator in Clinical Research |
|---|
| An IRB Coordinator manages ethical submissions, approvals, documentation, and communication with the Institutional Review Board, ensuring clinical trials follow ethical guidelines and protect participant rights throughout all trial phases. |
Behind every clinical trial that brings a new medicine, vaccine, or treatment to patients, there is a silent system working to protect human lives. Before a single participant is enrolled, before the first dose is administered, and before any data is collected, one critical question must be answered: Is this study ethically safe?
That responsibility lies with the Institutional Review Board (IRB) — an independent committee dedicated to safeguarding the rights, safety, and well-being of research participants. The IRB carefully examines clinical trial protocols, informed consent documents, recruitment strategies, and risk–benefit assessments to ensure that no participant is exposed to unnecessary harm. If ethical standards are not met, the study cannot proceed. Even after approval, the IRB continues to monitor the trial, ensuring ongoing compliance throughout its lifecycle.
But ethical decisions alone are not enough. They must be implemented, documented, tracked, and communicated effectively within the research environment. This is where the IRB Coordinator becomes essential. Acting as the bridge between investigators and the IRB, the IRB Coordinator manages submissions, maintains regulatory documentation, tracks approvals and renewals, and ensures that every ethical requirement and IRB ethical guidelines are consistently followed in daily research operations.
In simple terms, the IRB defines what is ethically acceptable, and the IRB Coordinator ensures that those standards are translated into action.
In this blog, we will explore how the IRB functions, how ethical review processes operate, what exactly an IRB Coordinator does, the skills required for this role, and why this career path plays such a vital role in modern clinical research.
| Who is an IRB Coordinator and Their Role in Clinical Research |
|---|
|
An IRB Coordinator is a clinical research professional responsible for organizing and managing the ethical review processes required for clinical trials involving human participants. In many institutions, this role functions similarly to an IRB administrator, supporting ethics committee operations and regulatory coordination. This role operates at the intersection of research operations and regulatory compliance. Their responsibilities include preparing and submitting ethics applications, tracking approval status, maintaining essential regulatory documentation, and facilitating communication between investigators and the Institutional Review Board (IRB). While the IRB evaluates study protocols, informed consent forms, recruitment materials, and risk–benefit assessments to determine whether a trial meets ethical standards, the IRB Coordinator ensures that these decisions are properly documented, communicated, and implemented throughout the lifecycle of the study. |
Clinical research cannot begin without prior approval from an independent ethics committee. International ethical frameworks such as the Declaration of Helsinki state that research involving human subjects must receive approval from an independent ethics committee before initiation (World Medical Association). Similarly, the ICH Good Clinical Practice (GCP) E6(R2) guideline requires that clinical trials undergo independent ethical review and continuous oversight to safeguard participant rights, safety, and well-being. The IRB Coordinator plays a critical role in operationalizing these requirements within the research environment.
Within the clinical research ecosystem, the IRB Coordinator holds a compliance-focused yet essential position. Unlike principal investigators or clinical research coordinators who manage trial execution and participant interactions, the IRB Coordinator ensures that ethical prerequisites remain valid at all times. This includes verifying that IRB approvals are active before study initiation, ensuring that the most recently approved informed consent versions are used, managing amendments and protocol modifications, and submitting continuing review documents within required timelines. By maintaining alignment with regulatory and ethical standards, the IRB Coordinator supports audit readiness and ensures that the study remains compliant throughout its duration.
In simple terms, the IRB defines what is ethically acceptable in a clinical trial, and the IRB Coordinator ensures that those ethical standards are consistently applied in daily research practice.
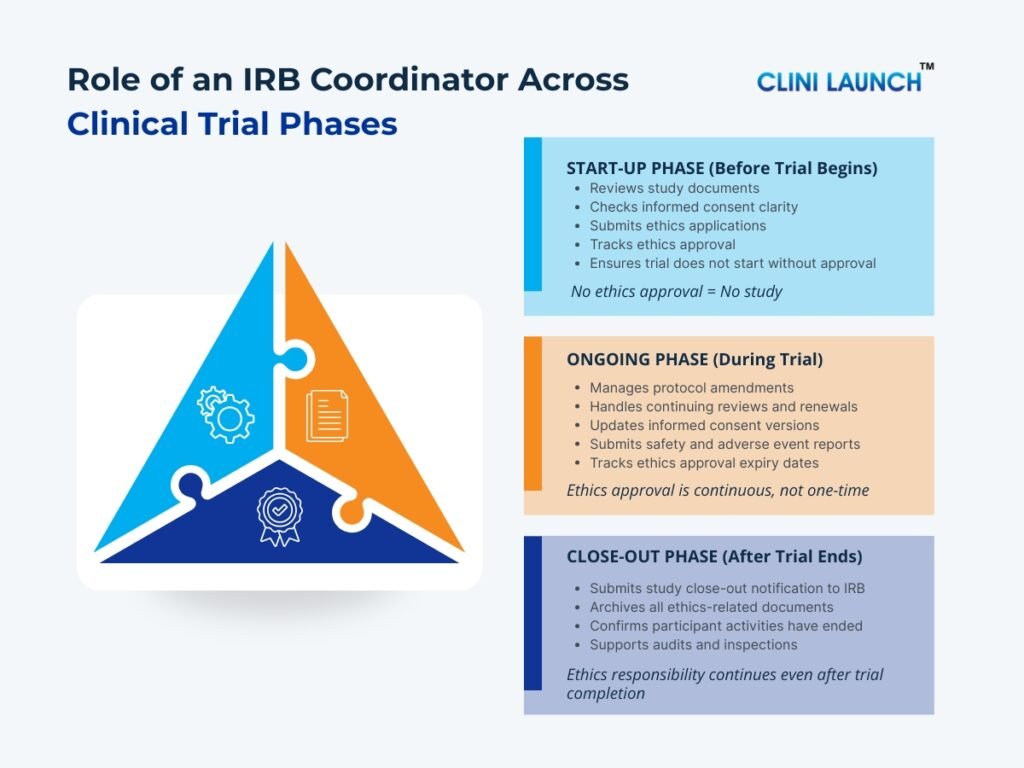
Roles and Responsibilities of an IRB Coordinator Across Clinical Trial Phases
The role of an IRB Coordinator is not limited to obtaining ethical approval at the beginning of a study. Their role spans the entire lifecycle of a clinical trial — from preparation before initiation, through ongoing study conduct, and finally to formal study closure. At every stage, the IRB Coordinator ensures that ethical standards are maintained, and regulatory requirements are fulfilled.
Prior to Commencement of the Clinical Trial (Start-Up Phase)
Before a clinical trial begins, ethical clearance must be secured from the Institutional Review Board (IRB). The IRB Coordinator plays a central role in preparing the study for ethical review and ensuring that no participant-related activities occur without proper authorization.
- Document Review and Preparation: The IRB Coordinator carefully reviews all study-related documents before submission, including the IRB research protocol, consent forms, recruitment materials, and investigator documentation. These typically include:
- Study protocol
- Informed consent forms (ICF)
- Recruitment materials
- Investigator brochure
- Safety monitoring plans
The coordinator ensures that documents are complete, internally consistent, and compliant with regulatory standards. Any missing information or inconsistencies are corrected before submission to prevent delays during IRB review.
- Ethical Submission and IRB Communication: Once documentation is finalized, the IRB Coordinator submits the complete ethics package to the IRB and manages the formal IRB submission process, ensuring that all required forms, supporting documents, and responses to committee queries are properly coordinated.
- . They serve as the primary liaison between the study team and the ethics committee, responding to queries, coordinating revisions, and implementing modifications requested by the IRB.
- Informed Consent Review: A critical responsibility during this phase is ensuring that the informed consent document clearly explains:
- Study purpose
- Procedures involved
- Potential risks and benefits
- Participant rights
- Voluntary participation
Consent forms must comply with regulatory requirements and ensure participants are fully informed before enrollment.
- Ethical Approval Tracking: The IRB Coordinator tracks approval status, maintains version control of documents, and records approval letters and conditions. Once approval is granted, they formally communicate the decision to investigators and confirm that study initiation may proceed.
- Preventing Premature Study Initiation: Before any participant is enrolled, the IRB Coordinator verifies that valid ethical approval is in place. They ensure that screening procedures, recruitment, and consent processes do not begin without formal IRB authorization. This safeguards participant safety and ensures strict ethical compliance.
| Case Study: Tuskegee Syphilis Study — Why Ethical Review Before Trial Initiation Is Essential |
|---|
|
In the 1930s, a clinical study in Tuskegee, Alabama, followed African American men with syphilis who were told they were receiving medical care, without being informed of the true purpose of the study. Participants were not given proper information, did not provide informed consent, and were denied effective treatment even after it became available. When the study was later exposed, it became a landmark example of unethical clinical research. It highlighted the serious risks of starting research without transparency, informed consent, and independent ethical oversight. This case led to strict requirements for ethical review before participant enrollment. Today, the IRB Coordinator’s primary responsibilities—such as document review, informed consent checks, ethical submissions, and confirmation of ethical approval—exist to prevent ethical failures like those seen in this study. |
During the Clinical Trial (Ongoing Phase)
Once a clinical trial begins, ethical oversight does not stop. The IRB Coordinator ensures continuous compliance throughout the study.
- Management of Protocol Amendments: During the study, changes to the protocol may be necessary due to safety findings or operational adjustments. The IRB Coordinator prepares and submits amendments to the IRB and ensures that no modifications are implemented before receiving ethical approval.
- Continuing Review and Renewal: IRB approvals are time-bound. The IRB Coordinator tracks approval of expiry dates and prepares continuing review submissions to ensure uninterrupted ethical authorization.
- Informed Consent Updates: If new safety information emerges, informed consent forms may need revision. The IRB Coordinator ensures updated versions are submitted for IRB approval and implemented correctly. Participants must be re-informed when necessary.
- Safety Reporting: The IRB Coordinator supports the submission of:
- Serious adverse events (SAEs)
- Safety updates
- Unanticipated problems
Timely reporting allows the IRB to reassess the risk–benefit balance and determine whether the study should continue.
- Monitoring Ethical Compliance: The coordinator ensures:
- Approved document versions are in use
- IRB recommendations are followed
- Expiry dates are monitored
- Regulatory documentation remains complete
By managing ethical-related activities during the ongoing phase, the IRB Coordinator ensures that the participant’s safety is continuously protected and that the clinical trial remains ethically and regulatory compliant until completion.
Multitasking Role of the IRB Coordinator in Multicenter Clinical Trials
In multicenter clinical trials, the complexity of ethical oversight increases significantly due to the involvement of multiple institutions, investigators, and review boards. Each participating site may have its own Institutional Review Board (IRB) processes, documentation standards, and communication requirements. This environment demands structured coordination to maintain consistency in ethical review, protocol implementation, and participant protection across all locations.
Research examining IRB processes in multicenter studies highlights the importance of structured liaison roles that facilitate communication between investigators and ethics committees, reduce review delays, and improve regulatory clarity (PubMed ID: 33367154). In such settings, the IRB Coordinator functions as a central coordinating figure.
The IRB Coordinator’s multitasking responsibilities include managing submissions across multiple sites, tracking varying approval timelines, coordinating amendments uniformly, ensuring that consent documents are harmonized while meeting local requirements, and maintaining version control across institutions. They must also monitor continuing reviews, safety reporting, and protocol compliance simultaneously for different centers.
In multicenter trials, ethical consistency is critical. The IRB Coordinator ensures that participant rights, safety, and regulatory standards are upheld uniformly, despite institutional differences. Their role extends beyond administrative processing — it becomes a coordination, compliance, and communication function essential for the ethical and efficient execution of large-scale clinical research.
| Case Study: Jesse Gelsinger Gene Therapy Trial — Why Ongoing Ethical Oversight Matters |
|---|
|
In 1999, Jesse Gelsinger, an 18-year-old participant, enrolled in a gene therapy clinical trial in the United States. During the trial, he experienced a severe immune reaction after receiving the investigational therapy and later died. Investigations revealed that serious adverse events from earlier participants were not fully reported, and certain protocol requirements were not strictly followed during the ongoing phase of the study. These gaps raised major concerns about safety monitoring, timely reporting to ethical committees, and adherence to approved study procedures. This case became a turning point in clinical research, highlighting that ethical approval at the start of a trial is not enough. Continuous oversight, timely safety reporting, protocol compliance, and ongoing ethical review are essential to protect participants. Today, the responsibilities handled by the IRB Coordinator during the ongoing phase—such as managing protocol amendments, tracking continuing reviews, updating informed consent, and ensuring timely safety reporting to the IRB—exist to prevent failures like those seen in this trial. |
After Trial Completion (Close-out Phase)
Even after participants complete their involvement in the study, ethical obligations continue. The close-out phase ensures that the clinical trial is formally concluded, with proper documentation and reporting to the Institutional Review Board.
- Study Close-out Notification to the IRB: Once the trial is completed or terminated, the IRB Coordinator submits a formal study close-out report to the IRB. This provides complete information to the IRB regarding the study activities involving the participants have been completed and that no further research procedures will take place.
- Final Documentation and Record Maintenance
The IRB Coordinator ensures that all ethical-related documents such as approval letters, consent forms, amendments, and safety reports are complete and properly archived. Maintaining accurate records is important for future audits, inspections, or regulatory reviews.
- Confirmation of End of Participant Involvement
The IRB Coordinator confirms that no participant-related activities continue after study closure. This helps to ensure that the participant’s rights and confidentiality remain protected even after the trial ends.
- Support During Audits or Inspections
If audits or inspections occur after the study completion, the IRB Coordinator supports the process by providing documentation and clarifications. This demonstrates that the study was conducted and closed in accordance with the ethical requirements.
By managing ethical-related activities during the close-out phase, the IRB Coordinator ensures that the study is ethically concluded and that all responsibilities towards the participants and regulatory bodies are fulfilled.
| Case Study: SUPPORT Trial — Importance of Ethical Oversight During and After Trial Conduct |
|---|
|
The SUPPORT trial (Surfactant, Positive Pressure, and Oxygenation Randomized Trial) was conducted to determine optimal oxygen levels for extremely premature infants. The study enrolled more than 1,300 newborns and compared outcomes for different oxygen saturation targets. Although the study had institutional ethical approvals, significant concerns were later raised about the informed consent process used in the trial. The U.S. Office for Human Research Protections (OHRP) identified that the informed consent forms did not adequately disclose reasonably foreseeable risks, such as possible death or serious complications related to differing oxygen levels—information that many ethicists and regulators argued should have been included. This led to a national debate on how risks should be communicated to participants or their guardians when clinical trials involve interventions within usual care but with varying risk levels. This controversy highlighted that ethical oversight must continue throughout the conduct of a study, not just at the beginning. Continuous ethical monitoring, transparent informed consent communication, and proper safety reporting—all managed by the IRB Coordinator during the ongoing and close-out phases—are essential to protect participant well-being. |
Difference Between IRB Coordinator and Clinical Research Coordinator (CRC)
The difference between an IRB Coordinator and a Clinical Research Coordinator arose because early clinical research did not clearly separate ethical oversight from trial execution, which led to serious ethical violations. When the same individuals in the operations of the trials were also involved in ethical monitoring process, participant safety was often compromised due to study timelines and operational pressure. To avoid this conflict of interest, clinical research systems were redesigned to keep ethical oversight independent from trial conduct.
How This Led to Two Distinct Roles
To protect the participants safety and ensure unbiased ethical decisions, responsibilities were divided:
- Ethical oversight and compliance were assigned to professionals supporting the Institutional Review Board, such as the IRB Coordinator.
- Day-to-day trial execution was assigned to site-based professionals, such as the Clinical Research Coordinator (CRC).
This separation ensures that ethical decisions are not influenced by trial targets, enrollment pressure, or operational challenges.
Key Differences Between IRB Coordinator and CRC
| Aspect | IRB Coordinator | Clinical Research Coordinator (CRC) |
|---|---|---|
| Primary Focus | Ethical review and compliance | Day-to-day conduct of the clinical trial |
| Main Responsibility | Manages ethical submissions, approvals, and documentation | Manages trial activities at the study site |
| Interaction with IRB | Works directly with the Institutional Review Board | Communicates with IRB mainly through the IRB Coordinator or sponsor |
| Interaction with Participants | No direct interaction with study participants | Direct interaction with participants (screening, visits, follow-ups) |
| Informed Consent Role | Reviews consent documents for ethical approval | Obtains informed consent from participants |
| Protocol Changes | Submits protocol amendments for ethical approval | Implements approved protocol changes at the site |
| Safety Reporting | Coordinates safety report submissions to IRB | Identifies and reports adverse events at the site |
| Study Phase Involvement | Involved across start-up, ongoing, and close-out phases | Involved mainly during trial conduct |
| Goal of the Role | Ensure participant rights, safety, and ethical compliance | Ensure smooth execution of the clinical trial |
Clinical Research
Build industry-ready skills to work across real clinical trial environments. Learn how clinical studies are designed, conducted, documented, and monitored, with a strong focus on ethical, patient safety, and global regulatory compliance.
Skills and Career Growth for an IRB Coordinator
An IRB Coordinator should have a basic understanding of the pros and cons of research with emphasis on the participants’ safety. in the clinical trials and support the work of the Institutional Review Board. The role mainly involves handling documents such as consent forms, ethical approvals, and study records, so being comfortable with paperwork and paying attention to small details is important. Clear communication skills are needed to coordinate with researchers and the ethical committee, explain the requirements, and follow up on approvals, while simple organizational skills help track submissions, approvals, and deadlines.
The IRB Coordinator role is often an entry or early-career position in clinical research ethical and compliance. With experience, professionals can grow into roles such as Senior IRB Coordinator or IRB Manager, or move into Regulatory Affairs, Compliance, Clinical Quality, or Research Governance positions. Since ethical oversight is mandatory for all clinical trials, professionals with IRB experience are consistently in demand. For individuals who prefer ethical, documentation, and compliance over patient-facing roles, this offers a stable and long-term career path in clinical research.
Conclusion
IRB Coordinators handle ethical paperwork and compliance for clinical trials to ensure participant safety and compliance.. Before the commencement of the trial review and documentation of the study protocols, consent forms, and recruitment materials, will be done and then submitted to the Institutional Review Board (IRB) for approval while tracking the process and communicating the updates to the research team. During the trial, they manage protocol changes by submitting amendments to the IRB, handle continuing reviews and renewals to keep approval active, update consent forms as needed, and forward safety reports like adverse events within the deadlines. After the trials end, IRB coordinator submits the close-out notifications, archives all ethical documents, and supports any audits to confirm full compliance.
Students interested in clinical research find this a practical entry-level role focused on the organization, documentation, and coordination skills with no direct patient interaction required. It builds experience across all trial phases (start-up, ongoing, and close-out), leading to growth in regulatory affairs, compliance management, or senior IRB positions. Enroll in an Advance Diploma in Clinical Research course at CliniLaunch to master these tasks and start a reliable career supporting ethical studies.
Frequently Asked Questions (FAQs) – IRB Coordinator
1. What does an IRB Coordinator do?
An IRB Coordinator manages ethical-related activities for clinical trials, including ethical submissions, approvals, documentation, and communication with the Institutional Review Board. Their role ensures that studies follow ethical guidelines throughout the trial lifecycle.
2. Is an IRB Coordinator a regulatory role?
Yes. An IRB Coordinator is considered part of ethical and regulatory compliance in clinical research, focusing on approvals, consent processes, and ethical oversight rather than trial execution.
3. Does an IRB Coordinator work with patients?
No. IRB Coordinators do not interact directly with patients. Their work is documentation- and compliance-focused, unlike roles such as Clinical Research Coordinator (CRC).
4. What qualifications are needed to become an IRB Coordinator?
Most IRB Coordinators come from life sciences, pharmacy, nursing, or clinical research backgrounds. Knowledge of research ethical and basic clinical research processes is important, while hands-on experience can be gained through the training programs.
5. Is IRB Coordinator a good role for freshers?
Yes. It is a good entry-level role for freshers who are interested in ethical, documentation, and compliance rather than patient-facing work.
6. What is the difference between IRB approval and ethical committee approval?
There is no major difference in purpose. “IRB” is commonly used in the U.S., while “Ethical Committee” is used in many other countries. Both exist to protect participant rights and safety.
7. Can a clinical trial start without IRB approval?
No clinical trial involving human participants can begin or continue without ethical (IRB) approval. Starting a trial without approval is a serious ethical and regulatory violation.
8. What skills are most important for an IRB Coordinator?
Attention to detail, basic understanding of research ethical, documentation skills, organization, and clear communication are the most important skills for this role.
9. What career growth is possible after working as an IRB Coordinator?
With experience, IRB Coordinators can grow into roles such as Senior IRB Coordinator, IRB Manager, Regulatory Affairs, Compliance, Clinical Quality, or Research Governance positions.
10. Is IRB Coordinator a stable long-term career?
Yes. Since ethical oversight is mandatory for all clinical trials, IRB and ethical-related roles are always in demand, making this a stable and long-term career option in clinical research.