Genomics Analyst
A genomics analyst is a life-science professional who analyzes DNA and RNA sequencing data to understand how genes function, change, and influence health and disease. They convert raw genetic data into meaningful biological insights using computers, data analysis, and bioinformatics tools rather than working primarily in wet labs.
Enquire About Bioinformatics TrainingImagine having an entire human genome in front of you and not knowing where to start. Millions of DNA reads, thousands of genes, and countless possible mutations sit inside a single dataset. This is the moment when genomics analysis truly begins. A genomics analyst doesn’t guess or scan randomly; they follow a precise sequence of tools to move from raw data to meaningful insight.
Genomics data analysis software is booming because next-generation sequencing (NGS) is now cheaper, and precision medicine is advancing fast. The market hit $1.68 billion in 2024. Pharma companies and research labs use these tools for combining data types and speeding up drug discovery.
Genomics data analysis is booming from $5.68 billion in 2024 to $20.49 billion by 2033 (15.4% CAGR), thanks to NGS advances and precision medicine. North America leads at 41.91% share, with pharma driving drug discovery growth.
Start Your Genomics Career with Practical Training
Who is a Genomics Analyst and What Do They Do?
A genomics analyst is a life-science professional who works with DNA and RNA sequencing data to understand genes, mutations, and their role in health and disease.
They perform quality checks, analyze NGS data (WGS, WES, RNA-seq), identify genetic variants, and interpret their biological significance. Their work supports clinical decisions, research, and drug development.
Genomics analysts also present findings through reports and visualizations, working across clinical labs, biotech companies, pharmaceutical organizations, and research institutions.
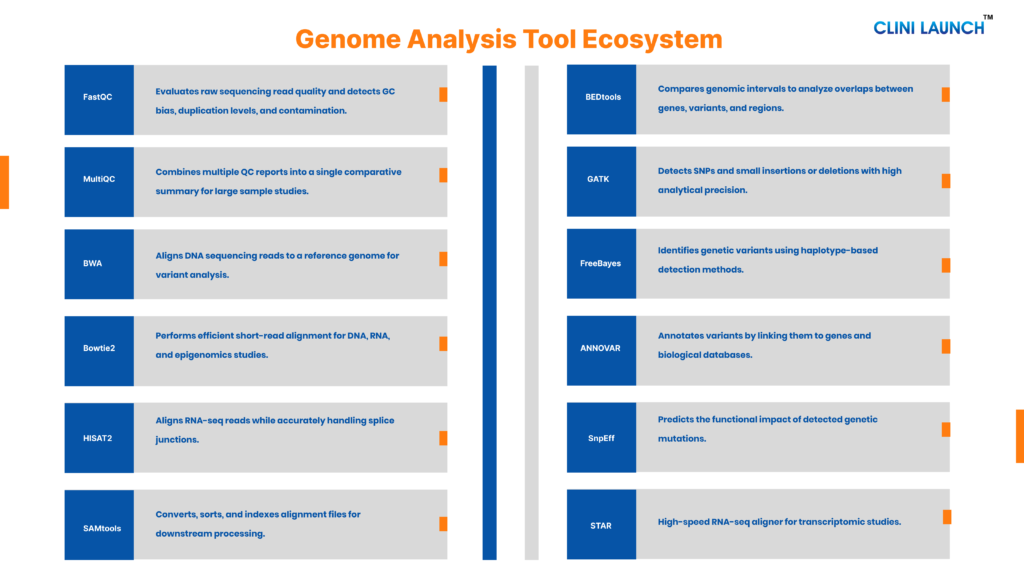
Top tools used by genomics analyst
Sequencing a genome produces massive amounts of data, but identifying meaningful patterns is complex. Genomics analysts use specialized tools to filter, analyze, and visualize this data to detect mutations and interpret biological significance.
Basic Tools Used by a Genome Analyst
These are foundational tools required in most standard DNA or RNA sequencing workflows. A genome analyst working in clinical genomics, research labs, or biotech will almost always use tools from these categories.
1.FastQC
FastQC is a widely used quality assessment tool designed for high-throughput sequencing data generated by next-generation sequencing (NGS) platforms. It is typically the first tool applied after raw FASTQ files are produced. Rather than modifying the dataset, FastQC performs a diagnostic evaluation of sequencing reads to identify technical biases, sequencing errors, and potential contamination. It generates standardized reports that help determine whether the data is suitable for downstream genomic analysis.
Genomic workflows such as alignment and variant calling depend heavily on data quality. Undetected issues like low-quality base scores, adapter contamination, or high duplication rates can compromise the accuracy of mutation detection and gene expression analysis. Running FastQC early prevents propagation of errors into later analytical stages.
Function
FastQC analyzes multiple quality metrics, including:
- Per-base sequence quality scores
- GC content distribution
- Sequence length distribution
- Adapter contamination detection
- Sequence duplication levels
- Overrepresented sequences
It produces graphical summaries that allow quick and systematic interpretation.
Skills Required
- Understanding of Phred quality scores
- Ability to interpret QC plots and warning flags
- Familiarity with FASTQ file format
- Basic Linux command-line proficiency
- Awareness of common sequencing artifacts
2. MultiQC
MultiQC is a reporting and aggregation tool used in genomics pipelines to consolidate outputs from multiple analysis tools into a single structured report. In large-scale sequencing projects involving many samples, individual quality control reports become difficult to compare manually. MultiQC streamlines this process by compiling results across samples and presenting them in a unified, comparative format. It does not perform primary analysis itself; instead, it enhances interpretability and standardization across datasets.
Large genomics studies require consistency across samples. Variability in sequencing depth, quality metrics, or contamination levels can affect downstream conclusions. MultiQC enables genome analysts to quickly detect sample outliers, batch effects, or systematic biases before moving forward with alignment and variant calling. This ensures reliability at the project level, not just at the individual sample level.
Function
MultiQC performs the following operations:
- Aggregates outputs from FastQC and other tools
- Summarizes metrics across multiple samples
- Generates comparative visual dashboards
- Highlights sample-level deviations
- Produces consolidated HTML reports
Skills Required
- Understanding of cohort-level sequencing metrics
- Ability to interpret aggregated QC summaries
- Familiarity with pipeline automation systems
- Basic command-line proficiency
- Awareness of batch-effect concepts
3. BWA (Burrows–Wheeler Aligner)
BWA is a widely adopted alignment tool used to map short DNA sequencing reads to a reference genome. It is primarily applied in whole genome sequencing (WGS) and whole exome sequencing (WES) workflows. BWA uses the Burrows–Wheeler Transform algorithm to index reference genomes efficiently, allowing rapid and memory-efficient alignment of millions to billions of sequencing reads. It forms the backbone of most DNA variant detection pipelines.
Accurate alignment is essential because every downstream step, including variant calling and structural analysis, depends on correct read placement. Misaligned reads can produce false-positive mutations or obscure real genetic variants. BWA ensures that reads are positioned correctly along chromosomal coordinates, creating the foundational BAM files required for further processing and mutation analysis.
Function
BWA performs the following operations:
- Indexes reference genome sequences
- Aligns short reads to genomic coordinates
- Handles mismatches and small gaps
- Generates SAM alignment output
- Supports paired-end read alignment
Skills Required
- Understanding of reference genome builds
- Familiarity with FASTA, FASTQ, SAM/BAM formats
- Knowledge of alignment scoring principles
- Linux command-line proficiency
- Awareness of mapping quality metrics
4. Bowtie2
Bowtie2 is a fast and memory-efficient alignment tool designed for mapping short sequencing reads to large reference genomes. It is commonly used in applications such as RNA sequencing, ChIP-seq, and epigenomics studies where high-throughput processing and flexible alignment parameters are required. Bowtie2 improves upon earlier short-read aligners by supporting gapped alignment, allowing it to handle insertions and deletions within reads more effectively.
Precise read alignment is critical for accurate downstream interpretation, particularly in studies where small mismatches or short indels can influence biological conclusions. Bowtie2 balances speed and sensitivity, making it suitable for large datasets that require efficient processing without excessive computational resource demands. It generates alignment files compatible with standard genomic workflows and integrates easily into automated pipelines.
Function
Bowtie2 performs the following operations:
- Builds indexed reference genomes
- Aligns short reads with gap support
- Handles mismatches and small indels
- Supports paired-end sequencing data
- Produces SAM output for downstream analysis
Skills Required
- Linux command-line proficiency
- Understanding of alignment parameters
- Knowledge of mismatch and gap penalties
- Familiarity with sequencing file formats
- Ability to interpret alignment statistics
5. HISAT2
HISAT2 is a splice-aware alignment tool specifically developed for RNA sequencing analysis. Unlike DNA aligners, it is optimized to handle reads that span exon exon junctions, which occur due to RNA splicing. HISAT2 uses a hierarchical indexing strategy that combines global and local genome indexing to achieve both speed and accuracy, even when working with large transcriptomic datasets. It is widely used in gene expression and transcript structure studies.
RNA-seq analysis requires specialized alignment because transcripts do not align continuously to the genome. Standard aligners may fail to correctly map spliced reads, leading to inaccurate gene expression results. HISAT2 accurately identifies splice sites and maps of reads across intronic regions, ensuring reliable quantification and downstream differential expression analysis.
Function
HISAT2 performs the following operations:
- Indexes reference genomes with splice site support
- Aligns RNA-seq reads across exon junctions
- Detects known and novel splice sites
- Supports paired-end sequencing
- Generates SAM alignment output
Skills Required
- Understanding of RNA biology and splicing
- Familiarity with gene annotation files (GTF/GFF)
- Linux command-line proficiency
- Knowledge of transcriptomics workflows
- Ability to interpret alignment metrics
Bioinformatics
Develop in-depth skills to analyze, manage, and interpret large-scale biological data used in genomics, clinical research, and drug discovery. This program focuses on applying computational methods and analytical pipelines to transform complex biological data into actionable research insights.
6. SAMtools
SAMtools is a command-line toolkit used for processing and managing alignment files generated after reading mapping. It works primarily with SAM (Sequence Alignment/Map) and BAM (Binary Alignment/Map) file formats, which store aligned sequencing reads along genomic coordinates. SAMtools is considered a foundational utility in genomics workflows because properly formatted and indexed alignment files are required before variant calling, visualization, or downstream statistical analysis can be performed.
After alignment, raw SAM files are typically large and inefficient for computation. SAMtools converts them into compressed BAM format, sorts read by genomic position, and indexes files for rapid access. Without these processing steps, variant detection tools cannot efficiently scan genomic regions. SAMtools ensure that alignment data is organized, accessible, and compatible with subsequent analysis stages.
Function
SAMtools performs the following operations:
- Converts SAM files to BAM format
- Sorts read by genomic coordinates
- Indexes BAM files for rapid querying
- Filters reads based on quality or flags
- Computes basic alignment statistics
Skills Required
- Understanding of SAM/BAM file structure
- Knowledge of genomic coordinate systems
- Linux command-line proficiency
- Familiarity with mapping quality scores
- Ability to manage large sequencing datasets
7. BEDtools
BEDtools is a powerful genomic analysis toolkit designed for comparing, intersecting, and manipulating genomic interval data. It operates on coordinate-based file formats such as BED, GFF, VCF, and BAM, allowing genome analysts to examine relationships between different genomic features. BEDtools are widely used in functional genomics to determine how variants, genes, regulatory elements, and sequencing peaks overlap within the genome.
Genomic data analysis often requires answering positional questions, such as whether a mutation falls within a gene, promoter region, or enhancer. BEDtools enable precise genomic arithmetic, allowing analysts to intersect variant coordinates with annotation datasets. This positional comparison is essential for interpreting biological significance, especially in regulatory and epigenomic studies.
Function
BEDtools performs the following operations:
- Intersects genomic intervals between datasets
- Identifies overlaps between variants and genes
- Calculates coverage across genomic regions
- Merges or subtracts genomic intervals
- Converts between coordinate-based file formats
Skills Required
- Understanding of genomic coordinate systems
- Familiarity with BED, GFF, and VCF formats
- Linux command-line proficiency
- Ability to interpret interval-based outputs
- Knowledge of gene annotation concepts
8. FreeBayes
FreeBayes is a haplotype-based variant calling tool used to detect genetic variations such as single nucleotide polymorphisms (SNPs), insertions, deletions, and complex polymorphisms from aligned sequencing data. Unlike position-based callers that analyze each genomic site independently, FreeBayes evaluates reads collectively to infer haplotypes, allowing more accurate detection of linked variants. It is commonly used in population genomics, non-model organisms, and multi-sample studies.
Accurate variant detection is central to genomic interpretation. FreeBayes supports multi-sample calling, enabling joint analysis across individuals to improve sensitivity and allele frequency estimation. This makes it particularly useful in cohort-based research and evolutionary studies. Its probabilistic framework allows flexible parameter tuning depending on sequencing depth and experimental design.
Function
FreeBayes performs the following operations:
- Detects SNPs and small indels
- Performs haplotype-based variant inference
- Supports multi-sample joint calling
- Generates VCF output files
- Estimates allele frequencies and genotype likelihoods
Skills Required
- Understanding of variant biology and haplotypes
- Familiarity with VCF file structure
- Knowledge of sequencing depth and coverage concepts
- Linux command-line proficiency
- Ability to interpret variant quality metrics
9. ANNOVAR
ANNOVAR is a widely used variant annotation tool that assigns functional and biological meaning to genetic variants identified during variant calling. After mutations are detected and stored in VCF format, ANNOVAR helps interpret their potential impact by mapping them to genes, transcripts, and external reference databases. It integrates genomic annotations with population frequency datasets and clinical repositories, enabling comprehensive downstream interpretation.
Variant detection alone does not explain biological significance. Many mutations may be benign, rare, or disease associated. ANNOVAR assists genome analysts in prioritizing variants by determining whether they fall within coding regions, alter amino acids, or appear in population databases at high frequency. This filtering and annotation step is essential in clinical genomics, rare disease research, and cancer genomics studies.
Function
ANNOVAR performs the following operations:
- Maps variants to genes and transcripts
- Classifies coding and non-coding mutations
- Integrates population frequency databases
- Retrieves functional prediction scores
- Generates annotated output tables
Skills Required
- Understanding of gene structure and mutation types
- Familiarity with annotation databases
- Ability to interpret functional prediction scores
- Knowledge of VCF format
- Basic command-line proficiency
10. SnpEff
SnpEff is a genetic variant annotation and effect prediction tool used to determine the potential biological impact of identified mutations. It analyzes variants in relation to annotated gene models and predicts how they may affect protein-coding sequences, splice sites, or regulatory regions. SnpEff is frequently integrated into variant calling pipelines to provide rapid functional categorization of mutations.
Understanding whether a variant causes an amino acid substitution, introduces a premature stop codon, or has no functional effect is critical for prioritization. SnpEff classifies variants into impact categories such as high, moderate, low, or modifier based on predicted consequences. This classification helps genome analysts filter large variant datasets and focus on mutations most likely to influence phenotypes or disease.
Function
SnpEff performs the following operations:
- Annotates variants relative to gene models
- Predicts coding and splice-site effects
- Classifies mutation impact levels
- Processes VCF files for downstream filtering
- Supports multiple genome annotation databases
Skills Required
- Understanding of coding sequence structure
- Knowledge of mutation impact categories
- Familiarity with VCF file format
- Ability to interpret transcript-level annotations
- Basic command-line proficiency
Advanced Tools Used by Genomic Analysts
These tools extend beyond foundational workflows. They are used in transcriptomics, structural genomics, single-cell studies, AI-driven modeling, and large-scale population projects.
11. DESeq2
DESeq2 is a statistical analysis package developed in R for identifying differentially expressed genes from RNA sequencing data. It operates on count-based data generated after reading alignment and quantification, using a negative binomial distribution model to estimate gene-level expression changes between experimental conditions. DESeq2 is widely applied in transcriptomics studies involving disease vs control comparisons, treatment response analysis, and biomarker discovery.
Gene expression datasets often contain variability due to sequencing depth and biological dispersion. DESeq2 addresses this by performing normalization, estimating variance across samples, and applying statistical testing to detect significant expression differences. It provides adjusted p-values to control false discovery rates, ensuring robust and reproducible results in high-dimensional datasets.
Function
DESeq2 performs the following operations:
- Normalizes raw count data
- Estimates dispersion parameters
- Conducts differential expression testing
- Calculates fold changes and adjusted p-values
- Generates summary statistics for gene-level analysis
Skills Required
- Proficiency in R programming
- Understanding of statistical modeling concepts
- Knowledge of RNA-seq count data structure
- Ability to interpret fold change and significance values
- Familiarity with data visualization in R
12. edgeR
edgeR is an R-based statistical package designed for differential expression analysis of count data derived from RNA sequencing experiments. It is particularly effective for experiments with small sample sizes or complex study designs. Like DESeq2, edgeR models count data using a negative binomial distribution, but it applies empirical Bayes methods to improve dispersion estimation across genes, enhancing statistical stability.
RNA-seq datasets often contain biological and technical variability that can obscure meaningful expression differences. edgeR accounts for this variability by estimating gene-wise dispersion and applying appropriate normalization strategies. It is well suited for multifactor experimental designs, time-series studies, and cases where replicates are limited. Its flexibility makes it a preferred tool in research settings requiring customized statistical modeling.
Function
edgeR performs the following operations:
- Normalizes sequencing count data
- Estimates common and gene-specific dispersion
- Conducts differential expression testing
- Supports multifactor experimental designs
- Outputs fold change and statistical significance metrics
Skills Required
- Proficiency in R programming
- Understanding of statistical inference concepts
- Knowledge of experimental design principles
- Ability to interpret dispersion estimates
- Familiarity with RNA-seq data preprocessing
13. STAR
STAR (Spliced Transcripts Alignment to a Reference) is a high-performance RNA sequencing alignment designed to map reads rapidly and accurately to a reference genome. It is specifically optimized for transcriptomic studies and is capable of handling very large datasets efficiently. STAR uses a sequential maximum mappable seed search approach, enabling precise detection of splice junctions while maintaining high computational speed.
RNA sequencing data presents unique challenges because transcripts contain spliced exons separated by introns in the genome. STAR addresses this by accurately aligning reads that span exon–exon boundaries, including detection of novel splice sites. Its speed and sensitivity make it suitable for large-scale gene expression studies and clinical transcriptomics projects where performance and accuracy are equally important.
Function
STAR performs the following operations:
- Indexes reference genomes for RNA alignment
- Maps read across exon–exon junctions
- Detects known and novel splice sites
- Supports paired-end sequencing
- Generates alignment output in SAM/BAM format
Skills Required
- Understanding of transcript structure and splicing
- Familiarity with RNA-seq workflows
- Linux command-line proficiency
- Knowledge of genome indexing procedures
- Ability to interpret alignment quality metrics
14. Manta
Manta is a structural variant detection tool designed to identify large-scale genomic alterations from next-generation sequencing data. Unlike SNP callers that focus on small nucleotide changes, Manta detects complex events such as insertions, deletions, inversions, duplications, and translocations. It analyzes paired-end and split-read alignment signals to infer structural rearrangements across the genome. Manta is commonly used in cancer genomics and germline studies where large chromosomal alterations play a critical biological role.
Structural variants can significantly impact gene function by disrupting coding regions or regulatory elements. Detecting these events requires algorithms capable of analyzing read orientation and breakpoint evidence. Manta integrates multiple alignment signals to improve sensitivity while maintaining specificity, making it suitable for both research and clinical workflows.
Function
Manta performs the following operations:
- Detects insertions, deletions, inversions, and translocations
- Analyzes paired-end and split-read evidence
- Identifies structural variant breakpoints
- Generates VCF output files
- Supports tumor-normal sample analysis
Skills Required
- Understanding of structural variant biology
- Knowledge of paired-end sequencing concepts
- Familiarity with VCF interpretation
- Linux command-line proficiency
- Ability to analyze breakpoint coordinates
15. CNVkit
CNVkit is a copy number variation (CNV) analysis tool used to detect genomic amplifications and deletions from targeted sequencing or whole-exome data. It evaluates read depth across genomic regions and compares coverage patterns between samples to identify copy number changes. CNVs are especially important in cancer genomics, where gene amplifications or deletions can drive disease progression.
Copy number alterations may not be visible through standard SNP or small indel detection tools. CNVkit processes aligned sequencing data to calculate coverage ratios and generate copy number profiles across chromosomes. It supports both tumor-only and tumor-normal comparative analyses.
Function
- Calculates read depth across genomic regions
- Detects copy number gains and losses
- Normalizes coverage using reference samples
- Generates CNV segmentation profiles
- Produces visualization-ready output
Skills Required
- Understanding of copy number biology
- Knowledge of sequencing coverage concepts
- Familiarity with BAM file processing
- Interpretation of chromosomal alteration plots
- Command-line proficiency
16. Seurat
Seurat is an R-based toolkit designed for single-cell RNA sequencing (scRNA-seq) analysis. It enables genome analysts to process, cluster, and interpret transcriptomic data at the individual cell level. Unlike bulk RNA-seq, single-cell analysis reveals heterogeneity within tissues and identifies distinct cell populations.
Seurat supports normalization, scaling, dimensionality reduction, clustering, and cell-type annotation. It is widely used in developmental biology, immunology, and cancer research to uncover cellular diversity.
Function
- Normalizes single-cell expression data
- Performs dimensionality reduction (PCA, UMAP)
- Identifies cell clusters
- Detects marker genes
- Visualizes cell population structure
Skills Required
- R programming proficiency
- Understanding of high-dimensional data
- Knowledge of clustering algorithms
- Interpretation of UMAP/t-SNE plots
- Statistical reasoning
17. Scanpy
Scanpy is a Python-based framework for scalable single-cell transcriptomics analysis. It is optimized for handling very large cell populations efficiently. Scanpy provides similar functionality to Seurat but integrates seamlessly with Python-based data science workflows.
It enables clustering, trajectory analysis, and visualization of gene expression patterns across thousands to millions of cells. Its scalability makes it suitable for large consortium-level projects.
Function
- Processes single-cell count matrices
- Performs clustering and trajectory analysis
- Executes dimensionality reduction
- Identifies differentially expressed genes
- Generates visualization plots
Skills Required
- Python programming
- Knowledge of matrix operations
- Understanding clustering and dimensionality reduction
- Interpretation of single-cell results
- Statistical analysis skills
18. IGV (Integrative Genomics Viewer)
IGV is a desktop-based genome visualization tool used for interactive inspection of aligned sequencing data. It allows genome analysts to examine read pileups, verify detected mutations, and visually confirm structural variants.
Automated pipelines may produce false-positive calls. IGV enables manual validation by displaying alignment patterns at specific genomic coordinates, helping to confirm variant authenticity.
Function
- Visualizes BAM and VCF files
- Displays read pileups
- Highlights variant positions
- Examines structural rearrangements
- Supports multiple annotation tracks
Skills Required
- Interpretation of alignment patterns
- Understanding read coverage visualization
- Knowledge of variant calling outputs
- Familiarity with genomic coordinates
- Analytical validation skills
19. UCSC Genome Browser (University of California, Santa Cruz Genome Browser)
The UCSC Genome Browser is a web-based genomic annotation platform provides access to reference genome assemblies and functional annotation tracks. It allows analysts to explore genes, regulatory elements, conservation scores, and known variants within genomic coordinates.
It is commonly used to contextualize detected variants and assess whether they lie in coding regions, promoters, enhancers, or conserved sequences.
Function
- Displays reference genome assemblies
- Integrates gene and regulatory tracks
- Visualizes conservation data
- Provides variant annotation context
- Supports coordinate-based search
Skills Required
- Understanding of gene structure
- Familiarity with genomic annotation tracks
- Ability to interpret regulatory elements
- Knowledge of reference genome builds
- Analytical interpretation skills
20. TensorFlow / PyTorch
TensorFlow and PyTorch are deep learning frameworks used for building neural network models in genomics. They enable predictive modeling for complex biological problems such as variant pathogenicity and gene expression prediction.
These frameworks are used when traditional statistical methods are insufficient for capturing nonlinear biological patterns.
Function
- Builds neural network architectures
- Trains predictive genomic models
- Processes high-dimensional biological data
- Supports GPU-accelerated computation
- Enables deep learning experimentation
Skills Required
- Advanced Python programming
- Understanding neural networks
- Knowledge of training and validation methods
- Experience with large datasets
- Model evaluation expertise
21. scikit-learn
scikit-learn is a Python machine learning library used for classification, regression, clustering, and dimensionality reduction tasks. In genomics, it is applied to predictive modeling and biomarker discovery.
It supports supervised and unsupervised learning algorithms suitable for genomic feature analysis.
Function
- Implements classification models
- Performs clustering
- Executes regression analysis
- Supports model evaluation
- Provides feature selection methods
Skills Required
- Python programming
- Understanding supervised learning
- Knowledge of evaluation metrics
- Feature engineering capability
- Statistical reasoning
22. AlphaFold-like Tools
AlphaFold-like systems predict protein three-dimensional structures from amino acid sequences using deep learning models. Structural prediction helps interpret how genetic mutations affect protein stability and function.
These tools bridge genomics and structural biology by linking sequence variation to functional consequences.
Function
- Predicts protein folding patterns
- Models structural conformations
- Analyzes mutation impact on structure
- Supports structural visualization outputs
Skills Required
- Understanding protein biology
- Familiarity with amino acid sequences
- Interpretation of structural models
- Basic computational modeling knowledge
23. Deep Variant
DeepVariant is an AI-based variant calling tool that uses deep neural networks to identify genetic variants from aligned sequencing data. It transforms sequencing information into image-like representations and applies deep learning classification.
It improves variant detection accuracy by reducing false positives and enhancing sensitivity.
Function
- Performs AI-driven variant calling
- Converts reads into image tensors
- Classifies SNPs and indels
- Outputs high-accuracy VCF files
Skills Required
- Understanding of variant calling workflows
- Knowledge of neural network basics
- Familiarity with BAM/VCF formats
- Computational resource management
Learn Top Genomics Analyst Tools Used in Industry
Challenges Faced by Genomic Analysts
Despite powerful computational tools, genome analysis remains a complex and demanding discipline. The challenges are not just technical they are analytical, biological, and infrastructural.
1. Explosive Data Volume
Whole-genome sequencing can generate hundreds of gigabytes per sample. Population-scale projects may involve thousands of genomes, pushing storage and computational infrastructure to their limits. Managing, transferring, and processing such datasets requires high-performance computing and optimized pipelines. Inefficient workflows can dramatically increase analysis time and cost.
2. Variant Interpretation Complexity
Identifying a mutation is straightforward compared to interpreting its biological significance. Many detected variants fall into the category of “variants of uncertain significance” (VUS). Determining whether a mutation is pathogenic, benign, or clinically actionable requires integration of databases, literature evidence, population frequency, and functional predictions. Interpretation remains one of the most intellectually demanding aspects of genomics.
3. False Positives and Technical Noise
Sequencing errors, alignment artifacts, and low coverage regions can produce misleading variant calls. Distinguishing true biological signals from technical artifacts requires cross-validation, visualization, and stringent filtering criteria.
4. Reproducibility and Pipeline Consistency
Genomics pipelines involve multiple tools, each with version dependencies and parameter configurations. Minor changes in software versions or filtering thresholds can alter results. Ensuring reproducibility across labs and studies is an ongoing challenge.
5. Multi-Omics Integration
Modern studies often combine genomics, transcriptomics, proteomics, and epigenomics data. Integrating heterogeneous datasets requires advanced computational frameworks and interdisciplinary expertise.
6. Ethical and Data Privacy Concerns
Genomic data is deeply personal. Secure storage, regulatory compliance, and controlled access are critical. Data misuse or breaches carry serious ethical and legal implications.
Future of Genomic Analysis Tools – The Role of AI
Genomic analysis tools are entering a new phase, driven largely by artificial intelligence (AI) and machine learning (ML). As genomic data volumes grow into the exabyte range, traditional methods are being supplemented or replaced by AI‑based systems that can detect hidden patterns, prioritize disease‑linked variants, and support clinical decisions at scale.
AI as a core analysis layer
The U.S. National Human Genome Research Institute (NHGRI) highlights that AI/ML is now central to interpreting large, complex genomic datasets from basic and clinical research. AI‑enabled tools are already used to distinguish disease‑causing variants from benign tumors, predict cancer progression, and improve the performance of gene‑editing tools such as CRISPR, making genome analysis more accurate and efficient.
Speed, scalability, and integration
AI speeds up multiple steps in genome analysis, from variant detection to outcome prediction, reducing manual review time, and enabling rapid re‑analysis of large cohorts. Government‑support initiatives such as NIH’s Bridge2AI program explicitly aim to embed AI into genomic and precision‑medicine workflows, emphasizing scalable, interoperable data pipelines and ethnically diverse datasets.
New generation of genome‑focused AI
Recent news coverage of DeepMind’s AlphaGenome notes that next‑generation AI models is designed to interpret long‑sequence variation and regulatory regions across the genome, offering fine‑grained predictions about variant impact in seconds. This kind of AI‑driven genome‑interpretation tool is being tested in rare‑disease and oncology research, where it can help solve previously undiagnosed cases and refine therapy selection.
Ethics, privacy, and future directions
Government‑led discussions also stress that AI‑augmented genome analysis must address privacy, bias, and data‑equity concerns. Going forward, genome‑analysis tools are expected to combine AI‑driven variant‑scoring, multi‑omics integration, and cloud‑scale infrastructure, turning AI from an add‑on into a foundational layer of genomic medicine.
Become a Job-Ready Genomics Analyst in 6 Months
Conclusion
For anyone aiming to become a genomics analyst, knowing these tools is not optional as it is essential to enter the field. Mastery of these tools demonstrates both technical proficiency and practical understanding, making candidates valuable in research laboratories, clinical genomics centers, and biotech companies. The more familiar you are with how these tools connect in real workflows, the more confident and job-ready you become.
At CliniLaunch Research Institute offers the Advanced Diploma in Bioinformatics designed to equip learners with hands-on skills and industry-relevant expertise. Enroll now to start building a successful career in genomics.
Frequently Asked Questions
Is coding mandatory to become a genome analyst?
Basic coding knowledge is strongly recommended, especially in R or Python. While some platforms provide graphical interfaces, most professional workflows require command-line and scripting skills for automation and scalability.
How long does it take to become proficient in genomics analysis tools?
With structured training and hands-on projects, foundational proficiency can be achieved in 6–12 months. Mastery develops through practical research or industry experience.
What is the difference between bioinformatics and genomics analysis?
Bioinformatics is a broader computational biology field, while genomics analysis focuses on DNA and RNA sequencing interpretation, variant detection, and gene-level insights.
Can genome analysts work outside healthcare?
Yes. Genome analysts also work in agriculture, evolutionary biology, microbiology, forensic science, and pharmaceutical research.
Do genome analysts work independently or in teams?
Most genome analysts work in interdisciplinary teams with molecular biologists, clinicians, statisticians, and data scientists.
What type of computing environment is typically used?
Genome analysis is commonly performed on Linux-based systems, high-performance computing clusters, or cloud platforms.
Are certifications necessary for a career in genomics?
Certifications are not mandatory but structured training and project experience significantly improve employability.
What industries are hiring genome analysts today?
Industries include precision medicine companies, cancer genomics labs, biotech startups, pharmaceutical firms, and population genomics programs.
How important is statistics in genome analysis?
Statistics is essential for sequencing data interpretation, expression analysis, and predictive modeling.
What career growth opportunities exist for genome analysts?
Professionals can advance to roles like senior bioinformatician, genomics scientist, computational biologist, AI-genomics specialist, or research lead.